

8. Complex formation involving double-bonded functional groups |
387 |
|||||||||||||||||||
|
|
C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ F2 |
|
|
|
|
|
|
|
|
|
C |
|
|
F |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
SCHEME 22 |
|
C |
|
|
F |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
C |
|
C |
|
|
|
|
|
|
|
|
|
+ |
|
|
F− |
|
C |
F |
||
|
|
|
|
|
|
|
|
|
|
C |
|
|
||||||||
+ F2 |
|
C |
F2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
C |
|
|
|
|
|
|
|
|
|
|
C |
F |
|
C |
F |
|||||
|
|
|
SCHEME 23 |
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
Ph |
Ph |
+ |
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
||||
CF3 OF |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
H |
H |
|
|
|
H |
|
OCF3 |
(enythro) |
|
|||||||||||
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
H |
F |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
Ph |
H |
+ |
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
CF3 OF |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
H |
Ph |
|
|
H |
|
OCF3 |
(threo) |
|
||||||||||||
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
F |
|
|
|
|||
SCHEME 24
(see Scheme 24). This tendency to cis addition may be explained by the presence of a short-lived carbocation (because of the unfavourable electron-withdrawing effect of the fluorine atom). The carbocation prefers the addition of a nucleophilic counter part (in a tight ion pair) to rotation around the C C bond.
H. Oxymercuration of Alkenes
Oxymercuration of alkenes is an electrophilic reaction involving the addition of mercuric salt and of a protic solvent (alcohols) according to reaction 9. UV/VIS spectrophotometric investigation of the olefin/mercuric salt reaction mixtures in methylene chloride provides evidence of the presence of an electron donor acceptor complex between olefin and mercuric salt110 which is considered to be on the reaction pathway of the oxymercuration.
C |
ROH |
RO |
|
C |
|
|
|
|
||
|
|
|
|
|
||||||
+ HgX2 |
|
|
|
|
|
|
|
+ HX |
(9) |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|||||
C |
|
|
|
|
C |
|
HgX |
|
||
|
|
|
|
|
|
|||||
The usual mechanism of oxymercuration111 is illustrated in Scheme 25. The presence of the mercuronium ion (53) was proposed by Winstein in 1939112.
388 |
Luciano Forlani |
|
C |
|
C |
+ |
HgX2 |
HgX2 |
C |
|
C |
|
|
(52) |
|
HgX |
|
|
+ |
X− |
|
C C |
|
(53)
+ ROH
HgX
|
|
+ HX |
C C |
||
OR
(54)
SCHEME 25
52 is the molecular complex which is in equilibrium with the starting reagents and with a CT complex 55 which is like that shown in equilibrium 10. The formation of 55 was
studied by 199Hg-NMR spectroscopy111.
C
K
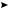 HgX2
HgX2
C
(52)
|
X |
|
|
|
C |
|
|
δ + |
Hg δ− |
|
(10) |
|
C |
|
|
|
|
|
|
|
X |
|
|
|
(55) |
|
|
|
|
|
It is clear that the mechanism in Scheme 25 parallels (at least from the qualitative point of view) the mechanism of the addition of bromine to olefins shown in Scheme 11. Kinetic investigations indicate that the oxymercuration reaction involves a series of fast equilibria until the mercuronium ion (53) is formed. The subsequent nucleophilic attack of the solvent is probably the rate-limiting step, as indicated by steric requirements in bulky alkenes111. In the bromine addition, the formation of the bromonium ion is the rate-limiting step (or the rate-limiting equilibrium). However, the olefin reactivities in both reactions (bromination and oxymercuration) are identical when steric effects in the TS of the two addition reactions are taken into account110.
I. Electrophilic Addition to Olefins: A Tentative Parallel with Other Reactions
In chemistry there is a tendency to label reactions in well ordered ‘pigeon holes’ with occasional interconnections. This is not surprising because often researchers in
8. Complex formation involving double-bonded functional groups |
389 |
various fields are anxious to obtain ‘taxonomical’ classifications. Consequently, attention is focused more on a single reaction or on some peculiarities of a reaction than on general principles. In addition, often the differences between reacting systems are emphasized, while the analogies are by-passed.
Probably, the electrophilic addition to olefins and the electrophilic aromatic substitution are more similar than is generally thought. Nevertheless, the two reactions are described in textbooks in separate chapters with the implicit idea that they are completely different reactions. Mechanistic investigations reveal that some steps show parallel behaviour in both reactions. In particular, the initial steps are similar for both reactions.
Many quantitative results regarding bromination of olefins and of aromatic substrates allow interesting comparisons to be made between the two reactions.
Strong differences in the reactivity of the aromatic CDC double bond compared to the reactivity of the CDC double bond of olefins are observed: olefinic electrophilic additions are faster than aromatic electrophilic substitutions. For instance, the addition of molecular bromine to cyclohexene (in acetic acid) is about 1014 times faster than the formation of bromobenzene from benzene and bromine in acetic acid113,114. Nevertheless, the addition of halogens to olefins parallels the Wheland intermediate formation in the halogenation of aromatic substrates.
The differentiation of the two kinds of reaction takes place in the step eliminating the positive charge from 27 and 28 in Scheme 10. This may occur by two main pathways:
(i)by neutralization of the positive charge by a nucleophile (which may be the counter ion of the electrophilic reagent or the solvent);
(ii)by elimination of positive charge with the removal of a proton, as usually observed in electrophilic aromatic substitution and in monomolecular elimination reactions of alkyl
halides.
In the case of particular olefins115, such as 1,2,3-tri-t-butylbutadiene (56), which is strongly sterically hindered, the usual dihalogenated products cannot be obtained. Instead of the addition process, the elimination of the positive charge by a proton removal takes
place73, as Scheme 26 illustrates. This reaction can be classified as an electrophilic substitution of the olefins.
+ Br2 |
−H+ |
|
|
+Br |
Br |
(56)
SCHEME 26
Electrophilic substitution reactions to olefins have been recorded on vinyl silanes116 (57). Scheme 27 reports the electrophilic substitution of the SiMe3 group by acyl halides. Probably, this reaction too occurs via a complex between the electrophilic part of the reagent and the system of the olefin, but other mechanisms are possible.
|
RCOCl |
SiMe3 |
COR |
(57)
SCHEME 27

390 |
Luciano Forlani |
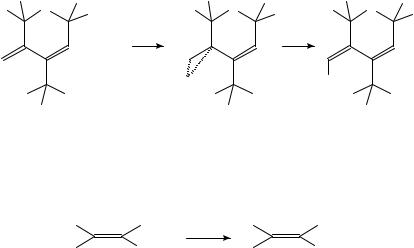
Elimination of the positive charge after the electrophilic attack on the olefin by proton removal is observed also in Scheme 28 between (E)-2,2,3,4,5,5-hexamethylhex-3-ene (58) and bromine. The CT complex shown in Scheme 28 probably turns into a second complex (with the ratio 1:2 of 58 and bromine) which ionizes to a bromonium/tribromide ion pair.
|
Br |
− |
|
|
3 |
|
|
|
Br2 |
H |
H |
|
|
|
C |
|
+ Br2 |
+ |
H |
|
|
||
|
|
Br |
|
(58) |
CT complex |
|
|
|
|
−H+ |
|
−HBr
CH3
Br
SCHEME 28
The theory that electrophilic aromatic substitution proceeds via a CT complex has been proposed for a long time in the literature117,118. In a recent paper, Kochi examines the relevance of the presence of a CT complex in electrophilic aromatic nitration119.
The generally accepted mechanism of the electrophilic aromatic substitutions120,121 involves the direct formation of the -complex (the Wheland intermediate) in the RDS (Scheme 29). The reversibility of the formation of the Wheland intermediate is under investigation. Many cases of reversibility of the steps shown in Scheme 29 have long been noted.
ArH + E+Y− |
slow |
H |
|
|
Ar + Y− |
|
ArE + HY |
||
|
|
|||
|
|
E |
|
|
|
|
Wheland |
|
|
E+ = electrophile |
complex |
|
|
|
|
|
|
||
Y− = ‘carrier’ |
|
|
|
|
|
|
SCHEME 29 |
|
|
As an alternative to the classical mechanism shown in Scheme 29, a multi-step mechanism involving an electron donor acceptor complex and a radical pair is shown in Scheme 30122. The distinction between the two mechanisms is difficult to make122 125. They can compete in forming the reaction products: the idea that only one reaction pathway is operating, is an oversimplification. However, two relevant points cannot be disregarded:
(i)In both reaction pathways, the formation of the substitution products arises from the-complex.
(ii)In the reaction mixtures, the presence of donor acceptor complexes is strongly borne out by spectroscopic evidence. Probably, these complexes are on the reaction pathways of the reactions under consideration.
8. Complex formation involving double-bonded functional groups |
391 |
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
ArH + E+Y− |
|
|
|
ArH E+ |
|
Y− |
|
|
|
ArH+.E . |
|
Y− |
|
|
|
Ar + |
Y− |
||
|
|
|
|
|
|
|
|
||||||||||||
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
CT complex |
|
|
radicalpair |
|
|
|
|
E |
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Wheland |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
complex |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ArE |
+ HY |
||||
SCHEME 30
Scheme 31 is a tentative collection of some reactions into a single picture with the purpose of emphasizing the common ‘segments’ of the reactions. Obviously, Scheme 31 includes some simplifications and assumptions (for instance, the nature of 60 may be questioned), and it may concern cyclic and non-cyclic as well as aromatic substrates.
E
(61)
|
|
−H+ |
|
|
|
|
Substitution |
+ E+ [Y−] |
E+ [Y−] |
+E [Y−] |
|
|
H |
H |
|
H |
(60) |
Y |
|
|
|
||
|
|
|
|
(59) |
+Z− |
|
E |
|
Sn1 |
||
|
|
|
H |
|
Z |
|
(62) |
|
E |
|
Addition |
(63)
SCHEME 31
The main reaction pathways indicated in Scheme 31 may be labelled as follows: 59 ! 62 (or 63): electrophilic addition to olefins.
59 ! 61: aromatic electrophilic substitution.
63 (or 62) ! 59 (or 61): monomolecular elimination.
63 ! 62 (and vice-versa): monomolecular nucleophilic substitution.
Y is the counter ion of the electrophilic reagent or of other nucleophiles present in the reaction mixtures, including neutral solvents.
392 Luciano Forlani
E is the electrophilic part of the halogen molecule. In the case of the simple addition of halogens, E D Y D F, Cl, Br, I.
Y may also be the leaving group in the monomolecular formation of substituted carbocations (60), to yield the saturated derivative 63 (and 63 may equal 62, if Z D Y), as usually indicated in SN1 reactions. If Z D E D Br, in the dibromo derivative 63, the cation 60 has the same form as that obtained by the bromination of olefins.
When Y D halogen (and E D H), 62 (and 63) are changed into olefin (61 or 59) by a monomolecular elimination reaction.
In aromatic electrophilic substitutions, 60 corresponds to the Wheland complex. Scheme 32 is a simplification of the Scheme 31.
k1 |
kx |
NUCLEOPHILE C ELECTROPHILE |
[INTERMEDIATE] ! PRODUCTS |
k |
|
1 |
|
SCHEME 32
The INTERMEDIATE may be obtained by a single equilibrium or by several subsequent equilibria, preceding the formation of the bond, such as that reported in equilibrium 11:
ELECTROPHILE |
|
K |
11 |
|
C |
NUCLEOPHILE MOLECULAR COMPLEX |
|||
|
|
|
|
|
The MOLECULAR COMPLEX may include all non-covalent interactions (donor |
|
accep- |
||
|
||||
tor, hydrogen bonding, . . .).
Scheme 32 is the one usually accepted for aromatic nucleophilic substitutions, as expected by considering the obvious idea that an electrophilic attack implies a simultaneous nucleophilic attack (and vice-versa). The conventional distinction between the reagent and the substrate is often related to the concentration ratio: the reagent is the one used in excess, while the substrate is the molecule which becomes modified.
In the reactions reported here the olefin is the nucleophile126. One obstacle to changing the conventional classification of reactions to a more appropriate and general expression arises also from the fact that usually the electrophilic reagent is a labile particle, often generated in situ and rarely in a known concentration.
By following an apparently correct assumption, as shown in Scheme 31, if the carbocation is formed in a RDS, all factors stabilizing 60 (solvent assistance, desolvating phenomena, delocalization of positive charge etc.) are factors enhancing the reaction rates of all pathways affording 60. On the other hand, the formation of 60 cannot be considered a simple RDS.
The generalization of Scheme 32 is valid when the products are obtained in a fast step (kx) from an intermediate obtained in a rate-limiting equilibrium (k1/k 1) (or in a series of equilibria). The fact that the intermediate cannot be obtained by irreversible reaction implies that the simple idea of the rate-limiting step127,128 cannot be used129: k1/k 1 is an equilibrium limiting the overall rate and the relative rates must be considered; k1 may be faster than kx, but if k 1 × k1 the ratio k1/k 1 is the factor which affects the overall reaction. The idea of the rate-limiting step becomes relatively insignificant when k 1 and k1 are of the same order of magnitude, and that is a frequent situation.
However, the formation of molecular complexes between nucleophile and electrophile (with a possible participation of solvents) in both electrophilic aromatic substitutions and nucleophilic aromatic substitutions is clearly expected, as shown by the conventional use of the terms ‘electrophile’ and ‘nucleophile’: these reactions belong to apparently different fields of organic chemistry, but the unification of the two kinds of reaction is mainly a matter of terminology and of details.

8. Complex formation involving double-bonded functional groups |
393 |
Actually, the collision between a discharged centre and a charged centre is the crucial event, whether in SNAr reactions or in electrophilic aromatic substitution reactions.
It is of interest to observe that the kinetic behaviour (and the effect of changes of temperature on the rate of reactions) of brominations of olefins and of electrophilic aromatic brominations confirms the presence of pre-associative processes77,130,131 on the reaction pathway, as well as that observed for some nucleophilic aromatic substitutions132.
An instance of an apparent electrophilic aromatic substitution (in this case 61 is an aromatic substrate, of Scheme 31), which actually is an electrophilic addition, is the halogenation of 2-aminothiazole derivatives which was usually considered a simple attack of the electrophilic reagent on the heterocyclic ‘aromatic’ substrate activated by the amino group; see reaction 12. When the bromination of 2-aminothiazole derivatives is carried out in nucleophilic solvents (ROH) and at low temperatures, the partially saturated derivatives (64) of Scheme 33 were isolated in 80 95% yields133. By heating 64, the usual halogenated 2-aminothiazoles are obtained, as indicated by Scheme 33. An apparent electrophilic aromatic substitution is actually an addition reaction to the CDC double bond; the elimination reaction is the following, separate step.
S |
|
|
|
|
X |
S |
|
|
|
NH2 + |
X2 |
|
|
|
NH2 |
HX |
|
|
|
|
||||||
N |
|
|
|
|
|
N |
(12) |
|
|
|
|
|
|
|
|||
X = Cl, Br |
|
|
|
|
|
|
||
|
|
|
Br |
H |
|
|
|
|
S |
|
|
S |
|
|
Br |
S |
|
|
|
|
|
|
||||
NH2 + |
Br2 |
ROH |
|
|
NH2 |
∆ |
|
NH2 |
|
|
|
− ROH |
|||||
N |
|
|
|
N |
|
N |
||
|
|
H |
|
|
|
|||
|
|
|
|
OR |
|
|
|
|
(64)
SCHEME 33
The fact that the partially saturated derivative (64) is obtained only in the trans form (these reactions are regioand sterespecific) indicates that the bromonium ion, 65, which is similar to that usually reported for halogenations of olefins, precedes the saturation of the CDC double bond.
H |
|
|
S |
Br + |
NH2 |
N
H
(65)
In the 1,3-thiazole system, the presence of sulphur is a factor stabilizing the bromonium ion, which may be obtained from a donor acceptor complex.
394 Luciano Forlani
J. Hydrogen Bonding Involving the p System of C=C Double Bond
It is known that bonds CDC of unsaturated compounds may act not only as electron donors toward metals, but also as proton acceptors for hydrogen bonding134.
Intramolecular and intermolecular interactions between C H groups and the system135 of carbon double bonds of olefins and of aromatic rings are of interest in the fields of molecular recognition and inclusion complexes in both organic chemistry and biochemistry.
IR and 1H-NMR investigations (as well as non-empirical and empirical calculations of interaction energies136) of facial interactions between hydroxy groups and CDC double bonds of olefins137,138 emphasize the importance of these interactions in explaining physical and chemical properties of molecules bearing double bonds and acid protons,
mainly of O H and N H groups. |
|
base shows |
139 |
11 alkenes (and |
||
Recent |
research on the Cambridge structural data |
|
||||
2 alkynes) which are involved in interactions with the |
OH group, with distance HÐ |
C |
||||
140Ð Ð |
||||||
of olefin |
˚ |
|
|
|
|
|
<2.4 A. 66 is an instance of intramolecular hydrogen bonding interactions . |
||||||
Intermolecular hydrogen bonds can be observed in 67141 and 68142. |
|
|
|
|||
|
|
|
H |
|
|
|
|
O |
O |
|
H |
|
|
|
H |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
HO |
|
|
|
|
(66) |
|
(67) |
|
|
|
|
|
OH |
OH |
|
|
|
|
|
|
O |
|
|
|
|
O |
H |
|
|
|
|
|
O |
O |
|
|
|
|
|
|
|
|
|
|
|
H
N
(68)
Even if hydrogen bonding with CDC double bonds can be hardly ascribed to a single carbon atom (or to electrons of the double bond), re-investigation of the structural data would be worthwhile because these hydrogen bonds were unrecognized in the original reports on the investigated structures.
The presence of HÐ Ð Ð bonding can relate to apparently anomalous reactivities. These interactions are of importance in chiral recognition and in other intermolecular and intramolecular interactions143.
8. Complex formation involving double-bonded functional groups |
395 |
Weak hydrogen-bonding interactions (the bond energy is in the range 1 2.2 kcal mol 1) between terminal alkynes and other alkynes or phenyl rings in the solid state were reported144. Hydrogen bonding with the system of a phenyl ring is revealed also by
|
|
|
-nitro-phenyl)hydrazone: an N |
|
H |
Ð Ð Ð |
interaction |
|||
X-ray diffraction of benzophenone (4145 |
|
|
|
|
˚ |
|||||
with the CDC bond of a phenyl ring |
is illustrated in 69, where a is 2.43 A and b |
|||||||||
˚ |
|
|
|
|
|
|
|
|
|
|
is 2.26 A. |
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
C a |
|
|
|
|
|
|
|
|
|
N |
H |
b |
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
N |
Me |
|
|
|
C |
|
|
Cl |
|
|
|
|
|
|
|
|
H |
H |
||
|
|
C |
a |
H |
|
|
|
|
||
|
|
C |
|
|
|
|||||
|
|
|
HCl |
|
|
|
|
|
||
|
|
|
|
|
|
C |
|
a |
||
|
|
C |
b |
C |
|
|
|
|||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
C |
|
|||
O2 N |
|
Me |
|
X H |
|
|
|
|
|
|
|
|
C |
|
|
|
|
|
H |
||
|
|
|
|
|
|
|
|
|
|
|
|
(69) |
(70) |
(71) |
|
|
|
(72) |
|
||
An on-face hydrogen bonding interaction between a hydroxy group of threonine and the-cloud of the aromatic ring of the tyrosine of isoenzyme 3-3 of glutathione S-transpherase was observed146. This interaction appears to act as a substituent bonded to a phenyl ring. In the solid state the HÐ Ð Ð distance between HCl proton and the two sp carbon atoms of 2-butyne (co-crystallized, 70) is 2.41 and 2.37 A˚ 147.
For intermolecular X HÐ Ð ÐC (in 71) hydrogen amine bonds (X D N) and of hydroxy groups (X D O), the mean obtained from data base analysis is 2.82 and 2.77 A,˚ respectively, for the C C bonds of olefins, and 2.61 and 2.69 A,˚ respectively, for the CDC triple bonds of alkynes. For the phenyl ring 2.47 and 2.43 A˚ (X D N and X D O, respectively) are calculated148.
The rotational spectrum of a hydrogen-bonded dimer formed by vinylacetylene and HCl has been recorded by the pulsed-nozzle FT microwave technique149. The structure of the complex 72 confirms the geometry of the van der Waals complexes predicted on the basis of simple electrostatic models150. The distance between the HCl proton and the
centre of the triple bond (a in 72) is 3.629 A;˚ the same distance with an isolated triple bond is 3.699 A˚ and with the double bond is 3.724 A˚ 150.
III. OLEFINS. COVALENT COMPLEXES WITH NUCLEOPHILES
Olefins are usually recorded as being unreactive toward nucleophilic reagents, unless electron-withdrawing groups are bonded to the carbon atoms adjacent to the carbon bearing the activating groups of CDC double bonds151. In fact, vinyl halides parallel the behaviour of aryl halides toward nucleophiles.
The attack of nucleophiles on activated CDC double bonds is an exciting field of research because the usual reactions of the CDC double bonds are reactions with electrophilic reagents152,153. Nitroalkenes react with nucleophiles by addition or substitution reactions that form interesting compounds for synthetic purposes: the nitro group is a versatile precursor for several groups154.
396 |
Luciano Forlani |
The usual substitution pathway for activated olefins is illustrated by Scheme 34 regarding anionic nucleophiles. In 73, R1 and R2 are electron-attracting groups; L is the leaving group (usually a halogen atom) and Y is the charged nucleophile (in particular, alkoxides and thioalkoxides).
Ar |
1 |
|
|
Y |
R1 |
|
|
|
|
Ar |
1 |
|
|||||
R |
|
|
|
|
|
|
|
|
|
|
R |
|
|||||
C C |
+ Y− |
|
Ar |
C |
C − |
|
|
|
|
|
|
|
|
C C |
pathway A |
||
|
−L− |
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
||||||||||||
L |
R2 |
|
|
|
|
|
|
|
Y |
|
R2 |
|
|||||
|
|
L |
2 |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
(73) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+H+ |
|
|
|
|
|
|
|
|
|
−HL |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Y |
|
|
R1 |
|
|
|
|||
|
|
|
|
|
Ar |
|
|
|
|
|
|
|
|
|
|
R2 |
|
|
|
|
|
|
|
C |
|
|
C |
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
pathway B |
|
|
|
|
|
|
|
|
|
L |
|
|
H |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
(74) |
|
|
|
|
|
|
|
SCHEME 34
Scheme 34 shows a two-step mechanism (pathway A), similar to the general addition/elimination mechanism of nucleophilic aromatic substitution reactions155. Pathway B of Scheme 34 shows the addition elimination mechanism. When nucleofuge expulsion is difficult (or the leaving group is absent) the saturated derivative 74 of Scheme 34 is the final product. Addition of amines to 1-methyl-4-vinylpyridinium cation (75) of reaction 13 is an instance of saturation of a CDC double bond by a nucleophilic attack156. R2NH of reaction 13 may be a primary or a secondary amine.
R2 NH + CH2 CH |
+ |
|
|
+ |
NCH3 |
|
R2 NCH2 CH2 |
NCH3 (13) |
|
|
(75)
The reactions between 1,1-dicyano-2-p-dimethylaminophenyl-2-fluoroethylene (76) and p-toluidine or phenylenediamines in acetonitrile157 form the substitution products (77) as shown in Scheme 35. The reaction is of the second order in amine when ArNH2 is p-toluidine and it is catalysed by substituted pyridines (and quinolines) in accordance with the presence on the reaction pathway of the zwitterionic complex 78, which is in equilibrium (see equation 14) with the anionic form 79.
|
Ar |
CN |
Ar |
CN |
||
+ |
− |
− |
||||
|
Base |
|
||||
ArNH2 |
C C |
|
|
ArNH C C |
(14) |
|
|
|
|||||
CN |
||||||
|
F |
F |
CN |
|||
|
|
|
|
|||
(78) |
(79) |