

Gale Encyclopedia of Genetic Disorder / Gale Encyclopedia of Genetic Disorders, Two Volume Set - Volume 1 - A-L - I
.pdfCleft palates are openings in the palate, which is the roof of the mouth. The size and position of the opening varies. The cleft may only be in the hard palate, the bony portion of the roof of the mouth opening into the floor of the nose, or it may only occur in the soft palate, the soft portion of the roof of the mouth. The cleft palate may involve both the hard and soft palate and may occur on both sides of the center of the palate.
Cleft lips can develop with or without cleft palates. Cleft palates may also occur without cleft lips.
Genetic profile
Cleft lip and palates not associated with a syndrome are caused by a combination of genetic and environmental factors. Inheritance caused by such a combination is called multifactorial. The embryo inherits genes that increase the risk for cleft lip and or palate. When an embryo with such genes is exposed to certain environmental factors, the embryo develops a cleft.
The risk of a baby being born with a cleft lip or palate increases with the number of affected relatives and increases with relatives that have more severe clefts.
Environmental factors that increase the risk of cleft lip and palate include cigarette and alcohol use during pregnancy. Some drugs also increase the incidence of clefting, such as phenytoin, sodium valproate, and methotrexate. The pregnant mother’s nutrition may affect the incidence of clefting as well.
Demographics
The incidence of cleft lip and palate not associated with a syndrome is one in 700 newborns. Native Americans have an incidence of 3.6 in 1,000 newborns. The incidence among Japanese newborns is two in 1,000. The incidence among caucasians is one in 1,000 newborns. African Americans have an incidence of 0.3 in 1,000 newborns.
Signs and symptoms
Babies born with a cleft lip will have an elongated opening in the upper lip. The size of this opening may range from a small notch in the upper lip to an opening that extends into the base of the nostril. The cleft lip may be below the right or left nostril or below both nostrils.
Babies born with a cleft palate will have an opening into the roof of the mouth. The size and position of the cleft varies and it may involve only the hard palate, or only the soft palate and may occur on both sides of the center of the palate.
An infant with a unilateral cleft lip. (Custom Medical Stock Photo, Inc.)
In some cases the cleft palate will be covered with the normal lining of the mouth and can only be felt by the examiner.
Infants with cleft lips and palates have feeding difficulties, which are more severe in those with cleft palates. The difficulty in feeding is due to the baby being unable to achieve complete suction. In the case of clefts of the hard palate, liquids enter the nose from the mouth through the opening in the hard palate.
A cleft palate also affects a child’s speech, since the palate is necessary for speech formation. The child’s speech pattern may still be affected despite surgical repair.
Ear infections are more common in babies born with cleft palates. The infections occur because the muscles of the palate do not open the Eustachian tubes which drain the middle ear. This allows fluid to collect and increases the risk of infection and hearing loss.
Teeth may also erupt misaligned.
palate and lip Cleft
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
241 |
Cleft lip and palate
Diagnosis
Cleft lip and palate can be diagnosed before birth by ultrasound. After birth, cleft lip and palate are diagnosed by physical exam.
Treatment and management
If cleft lip and/or palate are diagnosed by ultrasound before birth, further testing may be required to diagnose associated abnormalities if present. Referral to a cleft team is essential. A cleft team consists of specialists in the management of patients with clefts and includes surgeons as well as nurses and speech therapists. Members of the team inform the parents of all aspects of management. Feeding methods are also discussed, since feeding is the first problem that must be dealt with. It may be possible to breast feed a baby born with only a cleft lip, but babies born with cleft palates usually have more problems with feeding and frequently require special bottles and teats. A palatal obturator is a device that fits into the roof of the mouth, thus blocking the cleft opening and allowing easier suckling.
Surgery to repair cleft lips is sometimes performed after orthodontic treatment to narrow the gap in the upper lip. The orthodontic treatment can involve acrylic splints with or without screws or may involve the use of adhesive tape placed across the gap in the lip. Orthodontic treatment for cleft lip should begin within the first three weeks of life and continue until the cleft lip is repaired.
The timing of surgical cleft lip repair depends on the judgement of the surgeon who will perform the operation. The procedure is usually performed between one and three months of age. The goals of the operation are to close the gap in the upper lip, place scars in the natural skin curves, and to repair muscle so that the lip appears normal during movement. The closure is done in the three layers (skin, muscle, and mucosa) that line the inside of the lip. At the time of the procedure, if the nose is shaped abnormally due to the cleft lip, it is also corrected. Sometimes further surgery may be needed on the lip and or nose to refine the result.
The goals of the surgeon repairing a cleft palate are normal speech, normal facial growth, and hearing for the affected infant. The repair of the cleft palate is usually performed between three and 18 months of age. The timing may extend beyond this and varies with the type of cleft plate and center where the procedure is being performed. Depending on the type of cleft palate, more than one operation may be needed to close the cleft and improve speech.
Nonsurgical treatment of a cleft palate is available for patients who are at high risk for surgery and consists
of a prosthetic appliance worn to block the opening in the palate.
Babies born with cleft palates are vulnerable to ear infections. Their Eustachian tubes do not effectively drain fluid from the middle ear so fluid accumulates and infection sets in. This may lead to hearing loss. These children require drainage tubes to be inserted to prevent fluid accumulation.
Babies born with clefts usually require orthodontic treatment between 13 and 18 years of age. They also require speech therapy.
Prognosis
Individuals with cleft lip and palate have a good prognosis, and approximately 80% will develop normal speech. There is no known means of preventing clefting. Good prenatal care is essential and avoiding harmful substances appear to reduce the risk.
Resources
PERIODICALS
Bender, Patricia L. “Genetics of Cleft lip and Palate.” Journal of Pediatric Nursing 15 (August 2000): 242-249.
Christensen, Karr. “The 20th Century Danish Facial Cleft Population–Epidemiological and Genetic-Epidemiolical Studies.” Cleft Palate–Craniofacial Journal 36 (March 1999): 96-104.
Chung, Kevin C. “Maternal Cigarette Smoking during Pregnancy and the risk of having a child with Cleft Lip/Palate.” Plastic and Reconstructive Surgery 105 (February 2000): 458-491.
Cockell, Anna. “Prenatal Diagnosis and Management of Orofacial Clefts.” Prenatal Diagnosis 20 (February 2000): 149-151.
Denk, Michael J. “Topics in Pediatric Plastic Surgery.”
Pediatric Clinics of North America 45 (December 1998). Litwak-Saleh, Kim. “Practical Points in the case of the Patient
with post-Cleft lip repair.” Journal of Post Anesthesia Nursing 8 (February 1993): 35-37.
Rohrich, Rod J. “Optimal Timing of Cleft Palate Closure.”
Plastic and Reconstructive Surgery 106 (August 2000): 413-421.
ORGANIZATIONS
Cleft Palate Foundation. (800) 24-CLEFT.http://www.cleftline.org .
Farris F. Gulli, MD
Cleft palate see Cleft lip and palate
Cleidocranial dysostosis see Cleidocranial dysplasia
242 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

I Cleidocranial dysplasia
Definition
Cleidocranial dysplasia (CCD), also known as cleidocranial dysostosis, is a hereditary condition characterized by abnormal clavicles, delayed fusion of the bones in the skull, extra teeth, short stature, and other skeletal changes.
Description
Cleidocranial dysplasia is one of the skeletal dysplasia conditions, a large family of disorders involving abnormal growth and development of the skeleton.
CCD involves a characteristic group of abnormalities affecting primarily the skull, teeth, and clavicles. Other bones, such as the ribs, pelvis, and bones of the hands and feet may also be affected. Older children and adults with CCD are typically shorter than average. Most individuals with this condition do not have significant physical or mental disability.
Genetic profile
CCD is an autosomal dominant condition with variable expressivity (variable symptoms) and complete penetrance (meaning that all individuals who carry the gene for CCD have some symptoms). It is estimated that one third of cases represent new mutations, or genetic changes. The gene responsible for CCD has been mapped to the short arm of chromosome 6 and is called CBFA1. This gene encodes a transcription factor, meaning a protein that regulates DNA transcription, and is specifically expressed in the bone. Mutations in CBFA1 have been identified in many individuals and families with CCD.
Demographics
More than 500 cases of CCD among individuals of various ethnic backgrounds have been described in the medical literature. The incidence of CCD is reported to be highest around Cape Town, South Africa. The number of affected individuals in this area was estimated to exceed 1,000 as of 1996. These individuals descended from an affected Chinese sailor who settled in the area in 1896 and had seven wives. Study of this large family helped localize the gene responsible for the condition.
Signs and symptoms
Individuals with CCD typically show a delay or failure of the fusion of the calvarial sutures, the openings between the bones of the skull in infants. In some cases,
K E Y T E R M S
Clavicle—Also called the collarbone. Bone that articulates with the shoulder and the breast bone.
Deciduous teeth—The first set of teeth or “baby teeth”.
Fontanelle—One of several “soft spots” on the skull where the developing bones of the skull have yet to fuse.
Hypoplasia—Incomplete or underdevelopment of a tissue or organ.
Mutation—A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
the anterior fontanelle (the “soft spot” on an infant’s head) or other areas of the skull may remain unfused through life. A typical facial appearance in persons with CCD includes a broad forehead and widely spaced eyes. The overall head size is usually at the upper limit of normal.
Almost all persons with CCD have some degree of hypoplasia, or underdevelopment, of the clavicles (collar bones). In severe cases, both clavicles may be absent. More commonly, there is hypoplasia of the outside end of the clavicles. Depending on the degree of severity of clavicular hypoplasia, the external appearance of the shoulder may be affected. Some persons with CCD appear to have narrow, sloping shoulders, and some have the unusual ability to bring their shoulders together beneath their chin. This defect usually does not result in physical disability for the individual.
Dental abnormalities are very frequent among persons with CCD and are considered characteristic of the disorder. Almost all individuals are slow to lose their deciduous teeth (baby teeth), with a delay in the eruption of the permanent teeth. Some persons with CCD describe “living without teeth” until their permanent teeth started growing. Additionally, there may be a large number of extra teeth present. These extra teeth are so numerous so as to constitute a more or less complete third set of teeth. Additionally, the enamel of the teeth may be abnormal and prone to decay.
Other signs of CCD include a small rib cage with short or abnormal ribs. The vertebra of the spine may be malformed. The pelvis may be underdeveloped, with an increased space between the pubic bones. The growth of the bones in the hands and feet are often abnormal; most
dysplasia Cleidocranial
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
243 |

Cleidocranial dysplasia
This chest x ray shows the absence of collar bones, a feature common in cleidocranial dysplasia. (Greenwood Genetic Center)
are shorter but others are longer than normal. Final height in adults with CCD is usually shorter than expected given the family background.
More unusual complications associated with CCD include scoliosis (curvature of the spine), bone fragility, deafness, cleft palate, and a small jaw.
Diagnosis
The diagnosis of CCD is typically made by the doctor following review of the information obtained from physical exams, history, and x ray or other studies. The clavicular hypoplasia may only be seen on x rays.
The combination of hypoplastic clavicles, open fontanelles, and extra teeth is considered typical of CCD. The multiple dental anomalies in CCD are also quite specific and the diagnosis is evident in any individual with normal deciduous teeth, delayed eruption of permanent teeth, and multiple extra teeth.
Testing of the CBFA1 gene for mutations may also be performed. Identification of a mutation may confirm the initial diagnosis, or allow diagnosis before birth.
In a few cases, recognition of the features of CCD by ultrasound imaging, a technique that produces pictures of
the fetus, has led to diagnosis of the condition before birth.
Treatment and management
There is no specific treatment for cleidocranial dysplasia. Typically, a course of treatment is designed to manage the specific symptoms.
Children with CCD may be screened for deafness.
Long term dental treatment is often required. Surgery may be performed to remove the baby teeth and open the bony coverings surrounding the permanent teeth, with the goal of promoting their eruption. Orthodontic procedures may be required to align the teeth.
In pregnant females with CCD, the hypoplastic pelvis often necessitates a caesarian section delivery.
Prognosis
CCD is not expected to affect life expectancy in most cases and most diagnosed persons enjoy good overall health.
244 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

In some newborns, the small rib cage and reduced lung capacity may lead to respiratory distress. Height is often lower compared to that of other family members. The clavicular hypoplasia does not appear to significantly impair function, and some individuals with hypoplastic or absent clavicles have worked as manual laborers without difficulty. Dental problems are expected, and are sometimes severe enough so as to become a “dental disability”. Intelligence is usually normal.
Resources
BOOKS
Jones, K. L. Smith’s Recognizable Patterns of Human Malformation. W. B. Saunders Company, Philadelphia, 1997.
PERIODICALS
Mundlos, S. “Cleidocranial Dysplasia: Clinical and Molecular Genetics.” Journal of Medical Genetics 36 (1999):177-182.
Ramesar, Rajkumar S. et al. “Mapping the Gene for Cleidocranial Dysplasia in the historical Cape Town (Arnold) Kindred and Evidence for Locus Homogeneity.”
Journal of Medical Genetics 33 no. 6 (1996): 511-514.
Jennifer Roggenbuck, MS, CGC
I Clubfoot
Definition
Clubfoot is a condition in which one or both feet are twisted into an abnormal position at birth. The condition is also known as talipes.
Description
True clubfoot is characterized by abnormal bone formation in the foot. There are four variations of clubfoot, including talipes varus, talipes valgus, talipes equines, and talipes calcaneus. In talipes varus, the most common form of clubfoot, the foot generally turns inward so that the leg and foot look somewhat like the letter J. In talipes valgus, the foot rotates outward like the letter L. In talipes equinus, the foot points downward, similar to that of a toe dancer. In talipes calcaneus, the foot points upward, with the heel pointing down.
Clubfoot can affect one foot or both. Sometimes an infant’s feet appear abnormal at birth because of the intrauterine position of the fetus birth. If there is no anatomic abnormality of the bone, this is not true clubfoot, and the problem can usually be corrected by applying special braces or casts to straighten the foot.
K E Y T E R M S
Enterovirus—Any of a group of viruses that primarily affect the gastrointestinal tract.
Intrauterine—Situated or occuring in the uterus.
Orthopedist—A doctor specializing in treatment of the skeletal system and its associated muscles and joints.
Genetic profile
Experts do not agree on the precise cause of clubfoot. The exact genetic mechanism of inheritance has been extensively investigated using family studies and other epidemiological methods. As of 1999, no definitive conclusions had been reached, although a Mendelian pattern of inheritance is suspected. This may be due to the interaction of several different inheritance patterns, different patterns of development appearing as the same condition, or a complex interaction between genetic and environmental factors. The MSX1 gene has been associated with clubfoot in animal studies. But, as of 2001, these findings have not been replicated in humans.
A family history of clubfoot has been reported in 24.4% of families in a single study. These findings suggest the potential role of one or more genes being responsible for clubfoot.
Several environmental causes have been proposed for clubfoot. Obstetricians feel that intrauterine crowding causes clubfoot. This theory is supported by a significantly higher incidence of clubfoot among twins compared to singleton births. Intrauterine exposure to the drug, misoprostol, has been linked with clubfoot. Misoprostol is commonly used when trying, usually unsuccessfully, to induce abortion in Brazil and in other countries in South and Central America. Researchers in Norway have reported that males who are in the printing trades have significantly more offspring with clubfoot than men in other occupations. For unknown reasons, amniocentesis, a prenatal test, has also been associated with clubfoot. The infants of mothers who smoke during pregnancy have a greater chance of being born with clubfoot than are offspring of women who do not smoke.
Demographics
The ratio of males to females with clubfoot is 2.5 to 1. The incidence of clubfoot varies only slightly. In the United States, the incidence is approximately one in every 1,000 live births. A 1980 Danish study reported an overall incidence of 1.20 in every 1,000 children; by
Clubfoot
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
245 |

Clubfoot
A clubbed foot. (Photo Researchers, Inc.)
1994, that number had doubled to 2.41 in every 1,000 live births. No reason was offered for the increase.
Signs and symptoms
True clubfoot is usually obvious at birth. The four most common varieties have been described. A clubfoot has a typical appearance of pointing downward and being twisted inwards. Since the condition starts in the first trimester of pregnancy, the abnormality is quite well established at birth, and the foot is often very rigid. Uncorrected clubfoot in an adult causes only part of the foot, usually the outer edge, or the heel or the toes, to touch the ground. For a person with clubfoot, walking becomes difficult or impossible.
Diagnosis
True clubfoot is usually recognizable and obvious on physical examination. A routine x ray of the foot that shows the bones to be malformed or misaligned supplies a confirmed diagnosis of clubfoot. Ultrasonography is
not always useful in diagnosing the presence of clubfoot prior to the birth of a child.
Treatment and management
Most orthopedic surgeons agree that the initial treatment of congenital (present at birth) clubfoot should be non-operative. Non-surgical treatment should begin in the first days of life to take advantage of the favorable fibro-elastic properties of the foot’s connective tissues, those forming the ligaments, joint capsules, and tendons. In a common treatment, a series of casts is applied over a period of months to reposition the foot into a normal alignment. In mild cases, splinting and wearing braces at night may correct the abnormality.
When clubfoot is severe enough to require surgery, the condition is usually not completely correctable, although significant improvement is possible. In the most severe cases, surgery may be required, especially when the Achilles tendon, which joins the muscles in the calf to the bone of the heel, needs to be lengthened. Because an early operation induces fibrosis, a scarring and stiffness of the tissue, surgery should be delayed until an affected child is at least three months old.
Much of a clubfoot abnormality can be corrected by the use of manipulation and casting during the first three months of life. Proper manipulative techniques must be followed by applications of appropriately molded plaster casts to provide effective and safe correction of most varieties of clubfoot. Long-term care by an orthopedist is required after initial treatment to ensure that the correction of the abnormality is maintained. Exercises, corrective shoes, or nighttime splints may be needed until the child stops growing.
Prognosis
With prompt, expert treatment, clubfoot is usually correctable. Most individuals are able to wear regular shoes and lead active lives. If clubfoot is not appropriately treated, the abnormality becomes fixed. This has an effect on the growth of the leg and foot, and some degree of permanent disability usually results.
Resources
BOOKS
Hall, Judith G. “Chromosomal Clinical Abnormalilties.” In
Nelson Textbook of Pediatrics. 16th ed. Edited by Richard E. Behrman et al., 325–34. Philadelphia: Saunders, 2000.
Jones, KL. “XO Syndrome.” In Smith’s Recognizable Patterns of Human Malformation. 5th ed. Edited by Kenneth L. Jones and Judy Fletcher, 81–7. Philadelphia: Saunders, 1997.
Thoene, Jess G., ed. Physicians’ Guide to Rare Diseases. 2nd ed. Montvale, N.J.: Dowden Publishing Co., 1995.
246 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

Van Allen, Margot I., and Judith G. Hall. “Congenital Anomalies.” In Cecil Textbook of Medicine. 21st ed. Edited by Lee Goldman, et al., 150–52. Philadelphia: Saunders, 2000.
PERIODICALS
Chesney, D., et al. “Epidemiology and Genetic Theories in the Etiology of Congenital Talipes Equinovarus.” Bulletin of the Hospital for Joint Diseases 58, no. 1 (1999): 59–64.
Gonzalez, C. H., et al. “Congenital Abnormalities in Brazilian Children Associated with Misoprostol Misuse in First Trimester of Pregnancy.” Lancet 351, no. 9116 (May 30, 1998): 1624–27.
Honein, M. A., L. J. Paulozzi, and C. A. Moore. “Family History, Maternal Smoking, and Clubfoot: An Indication of a Gene-Environment Interaction.” American Journal of Epidemiology 157, no. 7 (October 1, 2000): 658–65.
Lochmiller, C., et al. “Genetic Epidemiology Study of Idiopathic Talipes Equinovarus.” American Journal of Medical Genetics 79, no. 2 (September 1, 1998): 90–6.
Rebbeck, T. R., et al. “A Single-Gene Explanation for the Probability of Having Idiopathic Talipes Equinovarus.”
American Journal of Human Genetics 53, no. 5 (November 1993): 1051–63.
Robertson, W.W., and D. Corbett. “Congenital Clubfoot. Month of Conception.” Clinics in Orthopedics 340, no. 338 (May 1997): 14–18.
ORGANIZATIONS
March of Dimes/Birth Defects Foundation. 1275 Mamaroneck Ave., White Plains, NY 10605. (888) 663-4637. resourcecenter@modimes.org. http://www.modimes
.org .
National Easter Seal Society. 230 W. Monroe St., Suite 1800, Chicago, IL 60606-4802. (312) 726-6200 or (800) 2216827. http://www.easter-seals.org .
National Organization for Rare Disorders (NORD). PO Box 8923, New Fairfield, CT 06812-8923. (203) 746-6518 or (800) 999-6673. Fax: (203) 746-6481. http://www
.rarediseases.org .
WEBSITES
“Clubfoot.” National Library of Medicine. http://www.nlm
.nih.gov/medlineplus/ency/article/001228.htm . Clubfoot.net. http://www.clubfoot.net/treatment.php3 . Ponseti, Ignacio, MD. “Treatment of Congenital Clubfoot.”
Revised January 1998. University of Iowa Health Care.
http://www.vh.org/Providers/Textbooks/Clubfoot/ Clubfoot.html .
Schopler, Steven A., MD. “Clubfoot.” Southern California Orthopedic Institute. http://www.scoi.com/clubfoot
.htm .
L. Fleming Fallon, Jr., MD, DrPH
Cobblestone dysplasia see Lissencephaly syndrome
I Cockayne syndrome
Definition
Cockayne syndrome (CS) is a rare inherited disorder that results in an extreme sensitivity to ultraviolet (UV) irradiation, mental retardation, and precocious (premature) aging.
Description
Since first reported in 1936 by Dr. Edward A. Cockayne, less than 200 cases of this disorder have been documented in medical literature. At birth, newborns with CS may have microcephaly (small-sized head) and low birthweight. During the first year of life they do not feed well and, as a result, they suffer from growth failure and delayed development. Ultimately, the disease usually results in death during the teenage years.
Genetic profile
CS results from mutations in the CSA gene (also known as the ERCC8 gene) located on chromosome 5. An affected person has inherited one abnormal or nonworking gene from each parent, a pattern that is consistent with autosomal recessive inheritance. When functioning normally, the CSA gene helps cells remove and destroy deoxyribonucleic acid (DNA) errors from strands undergoing active transcription. Also, the CSA gene allows cells to synthesize ribonucleic acid (RNA) after exposure to UV light. Although the parents of an affected child are normal, each of them carries an abnormal gene for CS. Therefore, they have a 25% risk with each pregnancy of having another affected child.
Demographics
CS occurs in less than one in 250,000 births and does not affect any one ethnic group more than another. Males and females are equally affected.
Signs and symptoms
The symptoms of CS are very striking. Failure to grow begins during the first year of life and results in the appearance of dwarfism. The patient’s weight is affected more than height. Also, some babies do not feed well and require feeding through a gastrostomy tube (a tube inserted through the abdominal wall into the stomach) to prevent malnutrition. As the infant grows, a delay in developmental milestones becomes apparent around the time that walking and talking should occur. Mental retardation in the mild to moderate range is found in all patients with CS. A small number of patients will have
syndrome Cockayne
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
247 |

Cockayne syndrome
K E Y T E R M S
Cataract—A clouding of the eye lens or its surrounding membrane that obstructs the passage of light resulting in blurry vision. Surgery may be performed to remove the cataract.
Contracture—A tightening of muscles that prevents normal movement of the associated limb or other body part.
Fibroblast—Cells that form connective tissue fibers like skin.
Gastrostomy—The construction of an artificial opening from the stomach through the abdominal wall to permit the intake of food.
Kyphosis—An abnormal outward curvature of the spine, with a hump at the upper back.
Microcephaly—An abnormally small head.
Mutation—A permanent change in the genetic material that may alter a trait or characteristic of an individual, or manifest as disease, and can be transmitted to offspring.
Myelin—A fatty sheath surrounding nerves in the peripheral nervous system, which help them conduct impulses more quickly.
Spasticity—Increased muscle tone, or stiffness, which leads to uncontrolled, awkward movements.
Transcription—The process by which genetic information on a strand of DNA is used to synthesize a strand of complementary RNA.
severe to profound mental retardation and some never have more than a few words of speech.
Other physical features include sun-sensitive skin, degeneration of retinal pigment, cataracts, and hearing loss. With exposure to sunlight, skin rashes appear and patients develop dry, scaly skin and thin hair. As part of the disease process, the skin develops an aged, leathery appearance. Although the eyes appear normal early in life, the retina later loses its pigment or color and develops a “salt-and-pepper” appearance. If cataracts appear within the first three years of life, the patient usually has the more severe form of CS that leads to death before adolescence. More than half the patients with CS have sensorineural hearing loss. The range of loss is from mild to severe.
Another finding of CS is an unusual gait (walk), caused by a combination of leg spasticity and contrac-
tures of the hips, knees, and ankles. The stooped posture often seen in CS results from kyphosis and joint contractures. Some of the first signs of neurologic changes are increased or decreased muscle tone and reflexes.
The most notable sign of CS is precocious senility (premature memory loss and confusion). Patients undergo neurological changes that resemble normal aging; the central and peripheral nervous systems lose myelin and neurons disappear from the central cortex and cerebellum. However, these changes occur at an extremely accelerated pace leading to death during early adolescence.
Diagnosis
Any child who displays these signs should have a genetic examination. CS is diagnosed by excluding other disorders. Specialized testing such as chromosome analysis, chromosome breakage studies, and DNA mutation analysis will rule out other genetic disorders such as
Bloom syndrome, Werner syndrome, and xeroderma pigmentosum. A person with CS will have a normal complement of 46 chromosomes. Their chromosomes also will not show any breakage when subjected to specialized laboratory analysis. DNA testing to look for the specific mutations in the CSA gene is also possible.
Only a very limited number of laboratories can perform the specialized testing that exposes cultured skin fibroblasts to UV irradiation. The fibroblasts of an affected person will lack the ability to form colonies.
Treatment and management
No specific treatment exists for CS. Patients should be treated according to the symptoms they have. Physical therapy will help prevent joint contractures that limit walking. Poor feeders may require a gastrostomy tube to prevent malnutrition. Patients should use sunscreen liberally and limit their exposure to sunlight. Special education will help to maximize the child’s learning potential.
Prognosis
The prognosis for CS is grim. Most patients die during the early adolescent years. Some survive until early adulthood. However, some patients have a more severe form and may die during early childhood.
Prevention
Since carriers of the gene that causes CS appear normal, and routine testing before pregnancy is not yet available, couples will not be aware of their risk until they
248 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |

have an affected child. For future pregnancies, prenatal diagnosis can determine whether or not the baby has CS.
Resources
PERIODICALS
Cleaver, J. E., et al. “A Summary of Mutations in the UV-sensi- tive Disorders: Xeroderma Pigmentosum, Cockayne Syndrome, and Trichothiodystrophy.” Human Mutation 14, no.1 (1999): 9-22.
Greenhaw, G. A., et al. “Xeroderma Pigmentosum and Cockayne Syndrome: Overlapping Clinical and Biochemical Phenotypes.” American Journal of Human Genetics 50, no.4 (April 1992): 677-89.
Higginbottom, M. C., et al. “The Cockayne Syndrome: An Evaluation of Hypertension and Studies of Renal Pathology.” Pediatrics 64, no. 6 (December 1979): 929-34.
Mathur, R., M. R. Chowdhury, and G. Singh. “Recent Advances in Chromosome Breakage Syndromes and Their Diagnosis.” Indian Pediatrics 37, no. 6 (June 2000): 615-25.
Nance, M. A., and S. A. Berry. “Cockayne Syndrome: Review of 140 cases.” American Journal of Medical Genetics 42, no. 1 (January 1, 1992): 68-84.
Sugita, T., et al. “Prenatal Diagnosis of Cockayne Syndrome Using Assay of Colony-forming Ability in Ultraviolet Light Irradiated Cells.” Clinical Genetics 22, no. 3 (September 1982): 137-42.
Suzanne M. Carter, MS, CGC
I Coffin-Lowry syndrome
Definition
Coffin-Lowry syndrome (CLS) is an inherited syndrome characterized by mental retardation, slow growth, distinctive facial appearance, large soft hands, loose joints, minor skeletal changes, and low muscle tone (hypotonia). Full expression of the disorder is seen only in males, although females may have some of the physical features and learning disability.
Description
Coffin-Lowry syndrome is one of a large number of mental retardation syndromes caused by abnormalities (mutations) of genes on the X chromosome. The pattern of physical findings, combined with mental retardation, makes the condition readily recognizable and its frequency makes it one of the well-known X-linked mental retardation syndromes. Although CLS was initially considered to be two separate syndromes, Coffin syndrome and Lowry syndrome, the two entities were recognized as the same disease in 1975.
K E Y T E R M S
Mental retardation—Significant impairment in intellectual function and adaptation in society. Usually associated an intelligence quotient (IQ) below 70.
X-linked—Located on the X chromosome, one of the sex chromosomes. X-linked genes follow a characteristic pattern of inheritance from one generation to the next.
Genetic profile
The gene for Coffin-Lowry syndrome, RSK2, is located on the short arm of the X chromosome designated as Xp22. Mutation of the RSK2 gene leads to full expression of the Coffin-Lowry syndrome in males since they only have a single X chromosome. If one of the two RSK2 genes is altered, it leads to some expression of the condition in the form of physical features and learning disabilities. Because females have two X chromosomes, CLS is considered inherited as an X-linked semidominant.
Demographics
Coffin-Lowry syndrome appears to occur in all populations. The full syndrome is seen in males with lesser expression in carrier females. A prevalence range of one in 50,000-100,000 males has been cited, but no studies with complete case findings have been conducted.
Signs and symptoms
Although the findings in Coffin-Lowry change with age, some manifestations are present from birth. Low muscle tone (hypotonia) and distinctive facial features that include prominent forehead, increased space between the eyes, forward direction of the nostrils, arching of the upper lip, and simple ear structure may be present in infancy. With the passing years, the face elongates, the ears become notably large, the lips and nasal structures thicken, and the mouth is usually open and agape. The hands are large and soft with thick fingers that narrow at their ends. There is generalized looseness at the joints. The central part of the chest may bow outward, the knees are flexed, and the feet flat.
Growth is slow, as manifest by low birth weight, a small head, and short stature during childhood and adult life. All developmental milestones in infancy and childhood are delayed, and intellectual function is severely impaired.
Milder findings consisting of short stature, increased space between the eyes, thick nasal tissues, prominent
syndrome Lowry-Coffin
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |
249 |
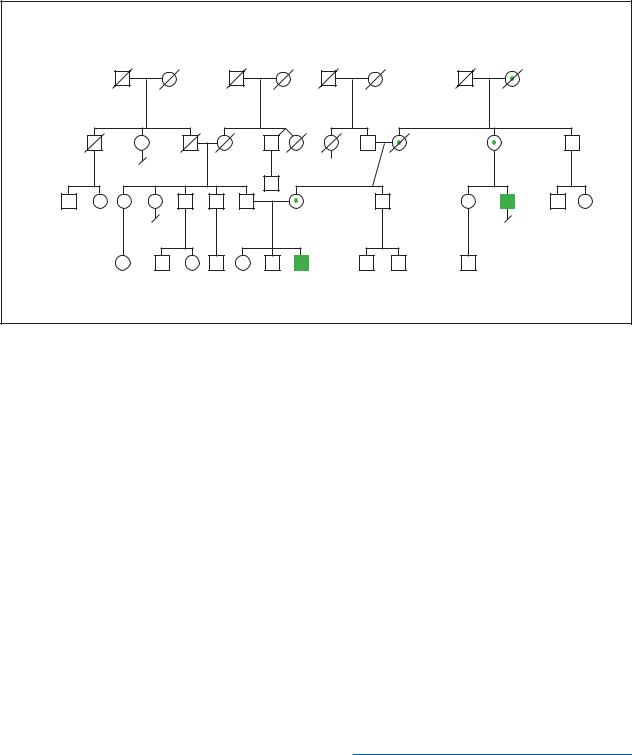
Coffin-Sirissyndrome |
Coffin-Lowry Syndrome |
|
|
|||
|
X-Linked Recessive |
|
|
|||
|
|
|
|
|
|
|
d. cancer |
|
|
d.1dy |
6' tall |
d. accident |
5' tall |
unknown |
|
|
? |
|
Broad nose |
Broad nose |
type |
|
|
|
|||
|
4 |
|
|
Psychiatric |
"Slow" |
|
|
|
|
|
|
||
2 |
|
|
5'1" tall |
|
problems |
|
|
|
|
|
|
||
|
43y |
|
40y Broad nose |
5'11" tall |
|
|
|
|
|
Tapered fingers |
|
Mental retardation |
|
|
|
|
Depression |
|
||
|
|
|
|
Deafness |
||
|
|
|
|
|
|
|
2 |
2 |
|
|
|
|
|
|
14y |
13y |
11y |
|
|
|
|
|
|
Mental retardation |
|
|
|
|
|
|
Deafness |
|
|
|
(Gale Group)
lips, and soft fleshy hands with thick fingers are consistently seen in carrier females. Intellectual function may be normal or mildly impaired.
Diagnosis
The diagnosis is usually based on the presence of the distinctive facial appearance and mental retardation. In many cases there will be a family history of other affected males or carrier females. X rays may show a number of minor features including delayed maturation of the bones, expansion at the ends of the bones of the digits, notching of the bones of the spine and narrowing of the space between the bones of the spine. The RSK2 gene responsible for Coffin-Lowry syndrome has been isolated, but gene testing is currently available only in research laboratories.
Treatment and management
There is no cure for Coffin-Lowry syndrome. There are no major malformations or specific health problems that pose complications. Because of severe mental retardation, lifelong supervision is generally required. Developmental progress can be promoted by early intervention, speech therapy, and physical therapy.
Prognosis
Long-term survival is the expectation, since individuals with Coffin-Lowry do not have any particular dis-
ease susceptibilities, nor do they have any major malformations. However, although there is an overall decrease in longevity in persons with severe mental retardation, specific information on survival in the Coffin-Lowry syndrome is not available.
Resources
PERIODICALS
Coffin, G. S., E. Siris, and L. C. Wegienka. “Mental Retardation with Osteocartilaginous Anomalies.” American Journal of
Diseases of Children 112 (1966): 205.
Lowry, B., and J. R. Miller. “A New Dominant Gene Mental Retardation Syndrome.” American Journal of Diseases of
Children 121 (1971): 496.
Temtamy, S. A., J. D. Miller, and I. Hussels-Maumenee. “The Coffin-Lowry Syndrome: An Inherited Faciodigital Mental Retardation Syndrome.” Pediatrics 86 (1975): 724.
Trivier, E., et al. “Mutations in the Kinase RSK-2 Associated with Coffin-Lowry Syndrome.” Nature 384 (1996): 567.
Roger E. Stevenson, MD
I Coffin-Siris syndrome
Definition
Coffin-Siris syndrome is a rare congenital disorder that affects more females than males. Individuals with this syndrome have some degree of mental retardation or
250 |
G A L E E N C Y C L O P E D I A O F G E N E T I C D I S O R D E R S |