

26510318-Bio-Transformation-of-Xenobiotics
.pdfCHAPTER 6 BIOTRANSFORMATION OF XENOBIOTICS |
173 |
sorbance maximum of cytochrome P450 is due to an unusual fifth ligand to the heme (a cysteine-thiolate). The amino acid sequence around the cysteine residue that forms the thiolate bond with the heme moiety is highly conserved in all P450 enzymes (Negishi et al., 1996). When this thiolate bond is disrupted, cytochrome P450 is converted to a catalytically inactive form called cytochrome P420. By competing with oxygen, CO inhibits cytochrome P450. The inhibitory effect of carbon monoxide can be reversed by irradiation with light at 450 nm, which photodissociates the cytochrome P450–CO complex.
These properties of cytochrome P450 are of historical importance. The observation that treatment of rats with certain chemicals, such as 3-methylcholanthrene, causes a shift in the peak absorbance of cytochrome P450 (from 450 to 448 nm) provided some of the earliest evidence for the existence of multiple forms of cytochrome P450 in liver microsomes. The conversion of cytochrome P450 to cytochrome P420 by detergents and phospholipases helped to establish the hemoprotein nature of cytochrome P450. The inhibition of cytochrome P450 by CO and the reversal of this inhibition by photodissociation of the cytochrome P450–CO complex established cytochrome P450 as the microsomal and mitochondrial enzyme involved in drug biotransformation and steroid biosynthesis (Omura, 1999).
The basic reaction catalyzed by cytochrome P450 is monooxygenation in which one atom of oxygen is incorporated into a substrate, designated RH, and the other is reduced to water with reducing equivalents derived from NADPH, as follows:
Substrate (RH) O2 NADPH H
Product (ROH) H2O NADP
Although cytochrome P450 functions as a monooxygenase, the products are not limited to alcohols and phenols due to rearrangement reactions (Guengerich, 1991). During catalysis, cytochrome P450 binds directly to the substrate and molecular oxygen, but it does not interact directly with NADPH or NADH. The mechanism by which cytochrome P450 receives electrons from NAD(P)H depends on the subcellular localization of cytochrome P450. In the endoplasmic reticulum, which is where most of the P450 enzymes involved in xenobiotic biotransformation are localized, electrons are relayed from NADPH to cytochrome P450 via a flavoprotein called NADPH–cytochrome P450 reductase. Within this flavoprotein, electrons are transferred from NADPH to cytochrome P450 via FMN and FAD. In mitochondria, which house many of the P450 enzymes involved in steroid hormone biosynthesis and vitamin D metabolism, electrons are transferred from NAD(P)H to cytochrome P450 via two proteins; an iron-sulfur protein called ferredoxin and an FMN-containing flavoprotein called ferredoxin reductase (these proteins are also known as adrenodoxin and adrenodoxin reductase). In bacteria such as Pseudomonas putida, electron flow is similar to that in mitochondria (NADH flavoprotein putidaredoxin P450).
There are some notable exceptions to the general rule that cytochrome P450 requires a second enzyme (i.e., a flavoprotein) for catalytic activity. One exception applies to two P450 enzymes involved in the conversion of arachidonic acid to eicosanoids, namely thromboxane synthase and prostacyclin synthase. These two P450 enzymes convert the endoperoxide PGH2 to thromboxane (TXA2) and prostacyclin (PGl2) in platelets and the endothelial lining of blood vessels, respectively. In both cases, cytochrome P450 func-
tions as an isomerase and catalyzes a rearrangement of the oxygen atoms introduced into arachidonic acid by cyclooxygenase. The plant cytochrome P450, allene oxide synthase, and certain invertebrate P450 enzymes also catalyze the rearrangement of oxidized chemicals. The second exception are two cytochrome P450 enzymes expressed in the bacterium Bacillus megaterium, which are known as BM-1 and BM-3 (or CYP106 and CYP102, respectively). These P450 enzymes are considerably larger than most P450 enzymes because they are linked directly to a flavoprotein. In other words, the P450 moiety and flavoprotein are expressed in a single protein encoded by a single gene. Through recombinant DNA techniques, mammalian P450 enzymes have been linked directly to NADPH–cytochrome P450 reductase and, like the bacterial enzyme, the resultant fusion protein is catalytically active. Most mammalian P450 enzymes are not synthesized as a single enzyme containing both the hemoprotein and flavoprotein moieties, but this arrangement is found in nitric oxide (NO) synthase. In addition to its atypical structure, the P450 enzyme expressed in B. megaterium, CYP102, is unusual for another reason: It is inducible by phenobarbital, as are some of the mammalian P450 enzymes (see “Induction of Cytochrome P450,” below).
Phospholipids and cytochrome b5 also play an important role in cytochrome P450 reactions. Cytochrome P450 and NADPH– cytochrome P450 reductase are embedded in the phospholipid bilayer of the endoplasmic reticulum, which facilitates their interaction. When the C-terminal region that anchors NADPH– cytochrome P450 reductase in the membrane is cleaved with trypsin, the truncated flavoprotein can no longer support cytochrome P450 reactions, although it is still capable of reducing cytochrome c and other soluble electron acceptors. The ability of phospholipids to facilitate the interaction between NADPH– cytochrome P450 reductase and cytochrome P450 does not appear to depend on the nature of the polar head group (serine, choline, inositol, ethanolamine), although certain P450 enzymes (those in the CYP3A subfamily) have a requirement for phospholipids containing unsaturated fatty acids.
Cytochrome b5 can donate the second of two electrons required by cytochrome P450. Although this would be expected simply to increase the rate of catalysis of cytochrome P450, cytochrome b5 can also increase the apparent affinity with which certain P450 enzymes bind their substrates; hence, cytochrome b5 can increase Vmax and/or decrease the apparent Km of cytochrome P450 reactions. In both cases, cytochrome b5 increases Vmax Km, which is a measure of catalytic efficiency and intrinsic clearance. Liver microsomes contain numerous forms of cytochrome P450 but only a single form of NADPH–cytochrome P450 reductase and cytochrome b5. For each molecule of NADPH–cytochrome P450 reductase in rat liver microsomes, there are 5 to 10 molecules of cytochrome b5 and 10 to 20 molecules of cytochrome P450. NADPH–cytochrome P450 reductase will reduce electron acceptors other than cytochrome P450, which enables this enzyme to be measured based on its ability to reduce cytochrome-c (which is why NADPH–cytochrome P450 reductase is often called NADPH–cytochrome-c reductase). NADPH–cytochrome P450 reductase can transfer electrons much faster than cytochrome P450 can use them, which more than likely accounts for the low ratio of NADPH–cytochrome P450 reductase to cytochrome P450 in liver microsomes. Low levels of NADPH–cytochrome P450 reductase may also be a safeguard to protect cells from the often deleterious one-electron reduction reactions catalyzed by this flavoprotein (see Fig. 6-13).

174 |
UNIT 2 DISPOSITION OF TOXICANTS |
The catalytic cycle of cytochrome P450 is shown in Fig. 6-35 (Dawson, 1988; Schlichting et al., 2000). The first part of the cycle involves the activation of oxygen, and the final part involves substrate oxidation, which entails the abstraction of a hydrogen atom or an electron from the substrate followed by oxygen rebound (radical recombination). Following the binding of substrate to the P450 enzyme, the heme iron is reduced from the ferric (Fe3 ) to the ferrous (Fe2 ) state by the addition of a single electron from NADPH–cytochrome P450 reductase. The reduction of cytochrome P450 is facilitated by substrate binding, possibly because binding of the substrate in the vicinity of the heme moiety converts the heme iron from a low-spin to a high-spin state. Oxygen binds to cytochrome P450 in its ferrous state, and the Fe2 O2 complex is converted to an Fe2 OOH complex by the addition of a proton (H ) and a second electron, which is derived from NADPH–cytochrome P450 reductase or cytochrome b5. Introduction of a second proton cleaves the Fe2 OOH complex to produce water and an (FeO)3 complex, which transfers its oxygen atom to the substrate. Release of the oxidized substrate returns cytochrome P450 to its initial state. If the catalytic cycle is interrupted (uncoupled) following introduction of the first electron, oxygen is released as superoxide anion (O2 ). If the cycle is interrupted after introduction of the second electron, oxygen is released as hydrogen peroxide (H2O2). The final oxygenating species, (FeO)3 , can be generated directly by the transfer of an oxygen atom from hydrogen peroxide and certain other hydroperoxides, a process known as the peroxide shunt. For this reason certain P450 reactions can be supported by hydroperoxides in the absence of NADPH–cytochrome P450 reductase and NADPH.
Figure 6-35. Catalytic cycle of cytochrome P450.
Cytochrome P450 catalyzes several types of oxidation reactions, including:
1.Hydroxylation of an aliphatic or aromatic carbon
2.Epoxidation of a double bond
3.Heteroatom (S-, N-, and I-) oxygenation and N-hydroxylation
4.Heteroatom (O-, S-, N- and Si-) dealkylation
5.Oxidative group transfer
6.Cleavage of esters
7.Dehydrogenation
In the first three cases, oxygen from the (FeO)3 complex is incorporated into the substrate, which otherwise remains intact. In the fourth case, oxygenation of the substrate is followed by a rearrangement reaction leading to cleavage of an amine (N-dealky- lation) or an ether (O- and S-dealkylation). Oxygen from the (FeO)3 complex is incorporated into the alkyl-leaving group, producing an aldehyde or ketone. In the fifth case, oxygenation of the substrate is followed by a rearrangement reaction leading to loss of a heteroatom (oxidative group transfer). The sixth case, the cleavage of esters, resembles heteroatom dealkylation in that the functional group is cleaved with incorporation of oxygen from the (FeO)3 complex into the leaving group, producing an aldehyde. In the seventh case, two hydrogens are abstracted from the substrate with the formation of a double bond (CPC, CPO, or CPN), with the reduction of oxygen from the (FeO)3 complex to water. It should be noted that this long list of reactions does not encompass all of the reactions catalyzed by cytochrome P450. As noted above (this section), cytochrome P450 can catalyze reductive reactions (such as azo reduction, nitro reduction, and reductive dehalogenation) and isomerization reactions (such as the conversion of PGH2 to thromboxane and prostacyclin). During the synthesis of steroid hormones, cytochrome P450 catalyzes the cleavage of carbon–carbon bonds, which occurs during the conversion of cholesterol to pregnenolone by side-chain cleavage enzyme (also known as P450scc and CYP11A1) and the aromatization of a substituted cyclohexane, which occurs during the conversion of androgens to estrogens by aromatase (also known as P450aro and CYP19).
Examples of aliphatic and aromatic hydroxylation reactions catalyzed by cytochrome P450 are shown in Figs. 6-36 and 6-37, respectively. The hydroxylation of aromatic hydrocarbons may proceed via an oxirane intermediate (i.e., an arene oxide) that isomerizes to the corresponding phenol. Alternatively aromatic hydroxylation can proceed by a mechanism known as direct insertion. The ortho-hydroxylation and para-hydroxylation of chlorobenzene proceed via 2,3- and 3,4-epoxidation, whereas meta-hydroxylation proceeds by direct insertion, as shown in Fig. 6-38. When aromatic hydroxylation involves direct insertion, hydrogen abstraction (i.e., cleavage of the C–H bond) is the rate-limiting step, so that substitution of hydrogen with deuterium or tritium considerably slows the hydroxylation reaction. This isotope effect is less marked when aromatic hydroxylation proceeds via an arene oxide intermediate. Arene oxides are electrophilic and therefore potentially toxic metabolites that are detoxified by such enzymes as epoxide hydrolase (see Fig. 6-6) and glutathione S-transferase. Depending on the ring substituents, the rearrangement of arene oxides to the corresponding phenol can lead to an intramolecular migration of a substituent (such as hydrogen or a halogen) from one carbon to the next. This intramolecular migration occurs at the site of oxidation

CHAPTER 6 BIOTRANSFORMATION OF XENOBIOTICS |
175 |
Figure 6-36. Examples of reactions catalyzed by cytochrome P450: Hydroxylation of aliphatic carbon.
and is known as the NIH shift—so named for its discovery at the National Institutes of Health.
Aliphatic hydroxylation involves insertion of oxygen into a C–H bond. As in the case of aromatic hydroxylation by direct insertion, cleavage of the C–H bond by hydrogen abstraction is the rate-limiting step, as shown below:
tochrome P450, a process variously known as metabolismdependent inhibition, suicide inactivation, or mechanism-based inhibition. As previously discussed in the section on epoxide hydrolase, not all epoxides are highly reactive electrophiles. Although the 3,4-epoxidation of coumarin produces an hepatotoxic metabolite, the 10,11-epoxidation of carbamazepine produces a stable, relatively nontoxic metabolite (Fig. 6-38).
(FeO)3 HC |
Fe(OH)3 ·C |
Fe3 HO C |
In the case of simple, straight-chain hydrocarbons, such as n- hexane, aliphatic hydroxylation occurs at both the terminal methyl groups and the internal methylene groups. In the case of fatty acids and their derivatives (i.e., eicosanoids such as prostaglandins and leukotrienes), aliphatic hydroxylation occurs at the -carbon (terminal methyl group) and the -carbon (penultimate carbon), as shown for lauric acid in Fig. 6-36. Most P450 enzymes preferentially catalyze the -1 hydroxylation of fatty acids and their derivatives, but one group of P450 enzymes (those encoded by the CYP4A genes) preferentially catalyzes the -hydroxylation of fatty acids, which can be further oxidized to dicarboxylic acids.
Xenobiotics containing a carbon–carbon double bond (i.e., alkenes) can be epoxidated (i.e., converted to an oxirane) in an analogous manner to the oxidation of aromatic compounds to arene oxides. Just as arene oxides can isomerize to phenols, so aliphatic epoxides can isomerize to the corresponding ene-ol, the formation of which may involve an intramolecular migration (NIH shift) of a substituent at the site of oxidation. Like arene oxides, aliphatic epoxides are also potentially toxic metabolites that are inactivated by other xenobiotic-metabolizing enzymes. Oxidation of some aliphatic alkenes and alkynes produces metabolites that are sufficiently reactive to bind covalently to the heme moiety of cy-
Figure 6-37. Examples of reactions catalyzed by cytochrome P450: Hydroxylation of aromatic carbon.

176 |
UNIT 2 DISPOSITION OF TOXICANTS |
Figure 6-38. Examples of reactions catalyzed by cytochrome P450: Epoxidation.
In the presence of NADPH and O2, liver microsomes catalyze the oxygenation of several S-containing xenobiotics, including chlorpromazine, cimetidine, lansoprazole, and omeprazole. Sulfurcontaining xenobiotics can potentially undergo two consecutive sulfoxidation reactions: one that converts the sulfide (S) to the sulfoxide (SO), which occurs during the sulfoxidation of chlorpromazine and cimetidine, and one that converts the sulfoxide (SO) to the sulfone (SO2), which occurs during the sulfoxidation of omeprazole and lansoprazole, as shown in Fig. 6-39. Albendazole is converted first to a sulfoxide and then to a sulfone. All of these reactions are catalyzed by FMO (as shown in Fig. 6-33B) and/or cytochrome P450 (as shown in Fig. 6-39). Both enzymes are efficient catalysts of S-oxygenation, and both contribute significantly to the sulfoxidation of various xenobiotics. For example, the sul-
foxidation of omeprazole, lansoprazole, chlorpromazine, and phenothiazine by human liver microsomes is primarily catalyzed by a P450 enzyme (namely CYP3A4), whereas the sulfoxidation of cimetidine and sulindac sulfide is primarily catalyzed by a flavin monooxygenase (namely FMO3). In the presence of NADPH and O2, liver microsomes catalyze the oxygenation of several N-containing xenobiotics — including chlorpromazine, doxylamine, oflaxacin, morphine, nicotine, MPTP, methapyrilene, methaqualone, metronidazole, pargyline, pyridine, senecionine, strychnine, trimethylamine, trimipramine, and verapamil—all of which are converted to stable N-oxides. Whereas S-oxygenation might be catalyzed by both cytochrome P450 and FMO, N-oxygenation is more likely to be catalyzed by just one of these enzymes. For example, the conversion of (S)-nicotine to trans-

CHAPTER 6 BIOTRANSFORMATION OF XENOBIOTICS |
177 |
Figure 6-39. Examples of reactions catalyzed by cytochrome P450: Heteroatom oxygenation.
(S)-nicotine N-1 -oxide by human liver microsomes is catalyzed by FMO3, with little or no contribution from cytochrome P450. Conversely, the conversion of pyridine to its N-oxide is primarily catalyzed by cytochrome P450. Both enzymes can participate in the N-oxygenation of certain xenobiotics. For example, the N-oxygenation of chlorpromazine is catalyzed by FMO3 and, to a lesser extent, by two P450 enzymes (CYP2D6 and CYP1A2). In general, FMO catalyzes the N-oxygenation of xenobiotics containing electron-deficient nitrogen atoms, whereas cytochrome P450 catalyzes the N-oxygenation of xenobiotics containing electron-rich nitrogen atoms. Therefore, substrates primarily N-oxygenated by cytochrome P450 are somewhat limited to pyri- dine-containing xenobiotics, such as the tobacco-specific nitrosamine NNK and the antihistamine temelastine, and to xenobiotics containing a quinoline or isoquinoline group, such as the muscle relaxant 6,7-dimethoxy-4-(4 -chlorobenzyl)isoquinoline. The initial step in heteroatom oxygenation by cytochrome P450 involves the abstraction of an electron from the heteroatom (N, S or I) by the (FeO)3 complex, as shown below for sulfoxidation.
(FeO)3 S |
(FeO)2 S |
Fe3 O S |
Abstraction of an electron from N, O, or S by the (FeO)3 complex is also the initial step in heteroatom dealkylation, but in this case abstraction of the electron from the heteroatom is quickly followed by abstraction of a proton (H ) from the -carbon atom
(the carbon atom attached to the heteroatom). Oxygen rebound leads to hydroxylation of the -carbon, which then rearranges to form the corresponding aldehyde or ketone with cleavage of the-carbon from the heteroatom, as shown below for the N-dealky- lation of an N-alkylamine:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(FeO)3 N |
|
|
|
|
|
|
|
(FeO)2 N |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH2R |
|
|
|
|
|
|
CH2R |
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
Fe(OH)3 N |
|
|
|
Fe3 N |
|
|
|
N |
|
O |
|
CHR |
|||||||||||
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
CHR |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
HOCHR |
|
H |
|
|
|
|
||||||||||
Although the initial steps in heteroatom oxygenation and heteroatom dealkylation are the same (abstraction of an electron from the heteroatom to produce a radical cation), the nature of the radical cation determines whether the xenobiotic will undergo oxygenation or dealkylation. The sulfur radical cations of numerous xenobiotics are sufficiently stable to allow oxygen rebound with the heteroatom itself, which results in S-oxygenation. However, this is not generally the case with nitrogen radical cations, which undergo rapid deprotonation at the -carbon, which in turn results in N-dealkylation. In general, therefore, cytochrome P450 catalyzes the N-dealkylation, not the N-oxygenation, of amines. N-Oxygenation by cytochrome P450 can occur if the nitrogen radical cation is stabilized by a nearby electron-donating group (mak-
178 |
UNIT 2 DISPOSITION OF TOXICANTS |
Figure 6-40. Examples of reactions catalyzed by cytochrome P450: Heteroatom dealkylation.
ing the nitrogen electron rich) or if -protons are either absent (e.g., aromatic amines) or inaccessible (e.g., quinidine). In the case of primary and secondary aromatic amines, N-oxygenation by cytochrome P450 usually results in the formation of hydroxylamines, as illustrated in Fig. 6-9. N-Hydroxylation of aromatic amines with subsequent conjugation with sulfate or acetate is one mechanism by which tumorigenic aromatic amines, such as 2-acetylaminoflu- orene, are converted to electrophilic reactive intermediates that bind covalently to DNA (Anders, 1985).
In contrast to cytochrome P450, which oxidizes nitrogencontaining xenobiotics by a radicaloid mechanism involving an initial one-electron oxidation of the heteroatom, the flavin monooxygenases oxidize nitrogen-containing xenobiotics by a heterolytic mechanism involving a two-electron oxidation by the 4ahydroperoxide of FAD (see Fig. 6-34). These different mechanisms explain why the N-oxygenation of xenobiotics by cytochrome P450 generally results in N-dealkylation, whereas N-oxygenation by the flavin monooxygenases results in N-oxide formation. In contrast to cytochrome P450, the flavin monooxygenases do not catalyze N-, O-, or S-dealkylation reactions.
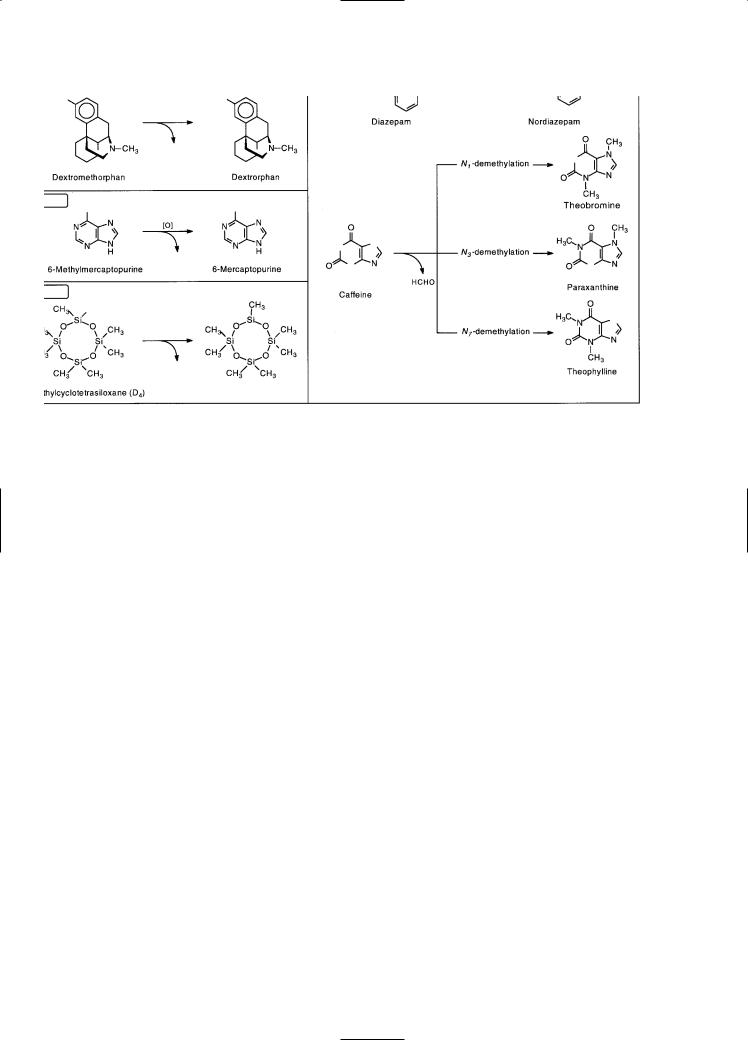
Numerous xenobiotics are N-, O-, or S-dealkylated by cytochrome P450, and some examples of these heteroatom dealkylation reactions are shown in Fig. 6-40. Cytochrome P450 has been shown to catalyze the demethylation of octamethylcyclotetrasiloxane, which is an example of Si-demethylation, as shown in Fig. 6-40. The dealkylation of xenobiotics containing an N-, O-, or S-methyl group results in the formation of formaldehyde, which can easily be measured by a simple colorimetric assay to monitor the demethylation of substrates in vitro. The expiration of 13C- or 14C-labeled carbon dioxide following the demethylation of drugs
Figure 6-41. Examples of reactions catalyzed by cytochrome P450: Oxidative group transfer.

CHAPTER 6 BIOTRANSFORMATION OF XENOBIOTICS |
179 |
containing a 13C- or 14C-labeled methyl group has been used to probe cytochrome P450 activity in vivo (Watkins, 1994). The activity of the human P450 enzymes involved in the N-demethyla- tion of aminopyrine, erythromycin, and caffeine can be assessed by this technique. Although caffeine has three N-methyl groups, all of which can be removed by cytochrome P450, the major pathway in humans involves N3-demethylation of caffeine to paraxanthine (see Fig. 6-40).
In addition to N-dealkylation, primary amines can also undergo oxidative deamination by cytochrome P450, which is an example of oxidative group transfer. The mechanism is similar to that of N-dealkylation: The -carbon adjacent to the primary amine is hydroxylated, which produces an unstable intermediate that re-
arranges to eliminate ammonia with the formation of an aldehyde or ketone. The conversion of amphetamine to phenylacetone is an example of oxidative deamination, as shown in Fig. 6-41. Oxidative deamination is also catalyzed by monoamine oxidase (MAO). In the example given above, however, the substrate, amphetamine, contains an -methyl group which renders it a poor substrate for MAO (as described earlier under “Monoamine Oxidase, Diamine Oxidase, and Polyamine Oxidase”).
In addition to oxidative deamination, cytochrome P450 catalyzes two other types of oxidative group transfer, namely oxidative desulfuration and oxidative dehalogenation. In all cases the heteroatom (N, S, or halogen) is replaced with oxygen. As shown in Fig. 6-42, oxidative desulfuration converts parathion, which has
Figure 6-42. Examples of reactions catalyzed by cytochrome P450: Cleavage of esters.

180 |
UNIT 2 DISPOSITION OF TOXICANTS |
little insecticidal activity, to paraoxon, which is a potent insecticide. The same reaction converts thiopental to pentobarbital. Diethyldithiocarbamate methyl ester, a metabolite of disulfiram, also undergoes oxidative desulfuration. The initial reaction involves S-oxidation by cytochrome P450 or FMO to a sulfine (R1R2CP S [O] R1R2C S OO ). In the presence of glutathione (GSH) and glutathione S-transferase, this sulfine is either converted back to the parent compound (R1R2CPS OO 2
GSH R1R2CPS GSSG H2O) or it undergoes desulfuration (R1R2CPS OO 2 GSH R1R2CPO GSSG H2S) (Madan et al., 1994).
Cytochrome P450 catalyzes both reductive and oxidative dehalogenation reactions (Guengerich, 1991). During oxidative dehalogenation, a halogen and hydrogen from the same carbon atom are replaced with oxygen (R1R2CHX R1R2CO) to produce an aldehyde or acylhalide, as shown in Fig. 6-16 for the conversion of halothane (CF3CHClBr) to trifluoroacetylchloride (CF3COCl). Oxidative dehalogenation does not involve a direct attack on the carbon–halogen bond, but it involves the formation of an unstable halohydrin by oxidation of the carbon atom bearing the halogen substituent. The carbon–halogen bond is broken during the rearrangement of the unstable halohydrin. When the carbon atom contains a single halogen, the resulting product is an aldehyde, which can be further oxidized to a carboxylic acid or reduced to a primary alcohol. When the carbon atom contains two halogens, the dihalohydrin intermediate rearranges to an acylhalide, which can be converted to the corresponding carboxylic acid (see Fig. 6-16). As discussed previously, aldehydes and, in particular, acylhalides are reactive compounds that can bind covalently to protein and other critical cellular molecules. The immune hepatitis caused by repeated exposure of humans to halothane and related volatile anesthetics is dependent on oxidative dehalogenation by cytochrome P450, with neoantigens produced by the trifluoroacetylation of proteins, as shown in Fig. 6-16.
As shown in Figs. 6-15 and 6-16, cytochrome P450 also can catalyze the reductive dehalogenation of halogenated alkanes and the reduction of certain azoand nitro-containing xenobiotics (Fig. 6-8). The ability of cytochrome P450 to reduce xenobiotics can be understood from the catalytic cycle shown in Fig. 6-35. Binding of a substrate to cytochrome P450 is followed by a oneelectron reduction by NADPH–cytochrome P450 reductase. Under aerobic conditions, reduction of the heme iron to the ferrous state permits binding of oxygen. Anaerobic conditions, in contrast, interrupt the cycle at this point, which allows cytochrome P450 to reduce those substrates capable of accepting an electron. Therefore, cytochrome P450 can catalyze reduction reactions, such as azo-reduction, nitro-reduction, and reductive dehalogenation, particularly under conditions of low oxygen tension. In effect, the substrate rather than molecular oxygen accepts electrons and is reduced. In fact, oxygen acts as an inhibitor of these reactions because it competes with the substrate for the reducing equivalents. The toxicity of many halogenated alkanes is dependent on their biotransformation by reductive dehalogenation. The first step in reductive dehalogenation is a one-electron reduction catalyzed by cytochrome P450, which produces a potentially toxic, carboncentered radical and inorganic halide. The conversion of CCl4 to a trichloromethyl radical and other toxic metabolites is shown in Fig. 6-15.
The oxidative desulfuration of parathion involves the production of an intermediate that rearranges to paraoxon (see Fig. 6-41). This same intermediate can decompose to para-nitrophenol and di-
ethylphosphorothioic acid, which are the same products formed by the hydrolysis of parathion (Fig. 6-42). In addition to facilitating the hydrolysis of phosphoric acid esters, cytochrome P450 also catalyzes the cleavage of carboxylic acid esters, as shown in Fig. 6-42. Carboxylic acid esters typically are cleaved by carboxylesterases, which results in the formation of an acid and an alcohol (R1COOCH2R2 H2O R1COOH R2CH2OH). In contrast, cytochrome P450 converts carboxylic acid esters to an acid plus aldehyde (R1COOCH2 R2 [O] R1COOH R2CHO), as shown in Fig. 6-42. The deacylation of loratadine is the major route of biotransformation of this nonsedating antihistamine. The reaction is catalyzed predominantly by cytochrome P450 (namely CYP3A4 with a minor contribution from CYP2D6), with little contribution from carboxylesterases.
Cytochrome P450 can also catalyze the dehydrogenation of a number of compounds, including acetaminophen, nifedipine, and related dihydropyridine calcium-channel blockers, sparteine, nicotine, valproic acid, digitoxin, and testosterone, as shown in Fig. 6-43. Dehydrogenation by cytochrome P450 converts acetaminophen to its hepatotoxic metabolite, N-acetylbenzo- quinoneimine, as shown in Fig. 6-28. Dehydrogenation of digitoxin (dt3) to 15 -dehydro-dt3 leads to cleavage of the terminal sugar residue to produce digitoxigenin bisdigitoxoside (dt2), which can similarly be converted to 9 -dehydro-dt2, which undergoes digitoxosyl cleavage to digitoxigenin monodigitoxoside (dt1). In contrast to digitoxin, this latter metabolite is an excellent substrate for glucuronidation. In rats, the P450 enzymes responsible for converting digitoxin to dt1 (namely the CYP3A enzymes) and the UDP-glucuronosyltransferase responsible for glucuronidating dt1 are inducible by dexamethasone, pregnenolone-16 -carbonitrile, and spironolactone, all of which protect rats from the toxic effects of digitoxin. The dehydrogenation of nicotine produces nicotine1 ,5 -iminium ion, which is oxidized by cytosolic aldehyde oxidase to cotinine, a major metabolite of nicotine excreted in the urine of cigarette smokers.
Testosterone is dehydrogenated by cytochrome P450 to two metabolites: 6-dehydrotestosterone, which involves formation of a carbon–carbon double bond, and androstenedione, which involves formation of a carbon–oxygen double bond. The conversion of testosterone to androstenedione is one of several cases where cytochrome P450 converts a primary or secondary alcohol to an aldehyde or ketone, respectively. The reaction can proceed by formation of a gem-diol (two hydroxyl groups on the same carbon atom), with subsequent dehydration to a keto group, as shown in Fig. 6-20 for the conversion of ethanol to acetaldehyde. However, gem-diols are not obligatory intermediates in the oxidation of alcohols by cytochrome P450, and in fact the conversion of testosterone to androstenedione by CYP2B1 (the major phenobarbitalinducible P450 enzyme in rats) does not involve the intermediacy of a gem-diol but proceeds by direct dehydrogenation (Fig. 6-43). In contrast, a gem-diol is involved in the formation of androstenedione from epi-testosterone (which is identical to testosterone except the hydroxyl group at C17 is in the -configuration, not the
-configuration). The fact that formation of androstenedione from epi-testosterone involves formation of a gem-diol, whereas its formation from testosterone does not, makes it difficult to generalize the mechanism by which cytochrome P450 converts alcohols to aldehydes and ketones.
Liver microsomes from all mammalian species contain numerous P450 enzymes, each with the potential to catalyze the various types of reactions shown in Figs. 6-36 through 6-43. In other

CHAPTER 6 BIOTRANSFORMATION OF XENOBIOTICS |
181 |
Figure 6-43. Examples of reactions catalyzed by cytochrome P450: Dehydrogenation.
words, all of the P450 enzymes expressed in liver microsomes have the potential to catalyze xenobiotic hydroxylation, epoxidation, dealkylation, oxygenation, dehydrogenation, and so forth. The broad and often overlapping substrate specificity of liver microsomal P450 enzymes precludes the possibility of naming these enzymes for the reactions they catalyze. The amino acid sequence of numerous P450 enzymes has been determined, largely by recombinant DNA techniques, and such sequences now form the basis for classifying and naming P450 enzymes (Gonzalez, 1989; Nelson et al., 1993). In general, P450 enzymes with less than 40 percent amino acid sequence identity are assigned to different gene families (gene families 1, 2, 3, 4, etc.). P450 enzymes that are 40 to 55 percent identical are assigned to different subfamilies (e.g., 2A, 2B, 2C, 2D, 2E, etc.). P450 enzymes that are more than 55 percent identical are classified as members of the same subfamily (e.g., 2A1, 2A2, 2A3, etc.). The liver microsomal P450 enzymes involved in xenobiotic biotransformation belong to three main P450 gene families, namely CYP1, CYP2, and CYP3. Liver microsomes also contain P450 enzymes encoded by the CYP4 gene family, substrates for which include several fatty acids and eicosanoids but relatively few xenobiotics. The liver microsomal P450 enzymes in each of these gene families belong to a single subfamily (i.e., CYP3A and CYP4A), two subfamilies (i.e., CYP1A and CYP1B) or five subfamilies (i.e., CYP2A, CYP2B, CYP2C, CYP2D, and
CYP2E). The number of P450 enzymes in each subfamily differs from one species to the next.
Human liver microsomes can contain 15 or more different P450 enzymes (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 3A4, 3A5, 3A7, 4A9, and 4A11) that biotransform xenobiotics and/or endogenous substrates (Guengerich, 1994; Wrighton and Stevens, 1992). Other P450 enzymes in human liver microsomes have been described, but they appear to be allelic variants of the aforementioned enzymes rather than distinct gene products. For example, CYP2C10 is an allelic variant of CYP2C9. Unfortunately, a nomenclature system based on structure does not guarantee that structurally related proteins in different species will perform the same function (examples of such functional differences are given later). Some P450 enzymes have the same name in all mammalian species, whereas others are named in a species-specific manner. For example, all mammalian species contain two P450 enzymes belonging to the CYP1A subfamily, and in all cases these are known as CYP1A1 and CYP1A2 because the function and regulation of these enzymes are highly conserved among mammalian species. The same is true of CYP1B1 and CYP2E1. In other words, CYP1A1, CYP1A2, CYP1B1, and CYP2E1 are not speciesspecific names but rather names given to proteins in all mammalian species. In all other cases, functional or evolutionary relationships are not immediately apparent; hence, the P450 enzymes are named

182 |
UNIT 2 DISPOSITION OF TOXICANTS |
in a species-specific manner and the names are assigned in chronological order regardless of the species of origin. For example, human liver microsomes express CYP2A6, but this is the only functional member of the CYP2A subfamily found in the human liver. The other members of this subfamily (i.e., CYP2A1 to CYP2A5) are the names given to rat and mouse proteins, which were sequenced before the human enzyme. With the exception of CYP1A1, CYP1A2, CYP1B1, and CYP2E1, the names of all of the other P450 enzymes in human liver microsomes refer specifically to human P450 enzymes.
Without exception, the levels and activity of each P450 enzyme have been shown to vary from one individual to the next, due to environmental and/or genetic factors (Meyer, 1994; Shimada et al., 1994). Decreased P450 enzyme activity can result from (1) a genetic mutation that either blocks the synthesis of a P450 enzyme or leads to the synthesis of a catalytically compromised or inactive enzyme, (2) exposure to an environmental factor (such as an infectious disease or a xenobiotic) that suppresses P450 enzyme expression, or (3) exposure to a xenobiotic that inhibits or inactivates a preexisting P450 enzyme. By inhibiting cytochrome P450, one drug can impair the biotransformation of another, which may lead to an exaggerated pharmacologic or toxicologic response to the second drug. In this regard, inhibition of cytochrome P450 mimics the effects of a genetic deficiency in P450 enzyme expression. Increased P450 enzyme activity can result from (1) gene duplication leading to over expression of a P450 enzyme; (2) exposure to environmental factors, such as xenobiotics, that induce the synthesis of cytochrome P450; or (3) stimulation of preexisting enzyme by a xenobiotic.
Although activation of cytochrome P450 has been documented in vitro, it appears to occur in vivo only under special circumstances. Although duplication of functional P450 genes has been documented, induction of cytochrome P450 by xenobiotics is the most common mechanism by which P450 enzyme activity is increased. By inducing cytochrome P450, one drug can stimulate the metabolism of a second drug and thereby decrease or ameliorate its therapeutic effect. A dramatic effect of this type of drug interaction is the induction of ethinylestradiol metabolism by phenobarbital and rifampin, which can ameliorate the contraceptive effect of the former drug and lead to unplanned pregnancy. Allelic variants, which arise by point mutations in the wild-type gene, are another source of interindividual variation in P450 activity. Amino acid substitutions can increase or, more commonly, decrease P450 enzyme activity, although the effect may be substrate-dependent. Examples of genetic factors that influence P450 activity are given later in this section. The environmental factors known to affect P450 levels include medications (e.g., barbiturates, anticonvulsants, rifampin, troglitazone, isoniazid), foods (e.g., cruciferous vegetables, charcoal-broiled beef), social habits (e.g., alcohol consumption, cigarette smoking), and disease status (diabetes, inflammation, viral and bacterial infection, hyperthyroidism, and hypothyroidism). When environmental factors influence P450 enzyme levels, considerable variation may be observed during repeated measures of xenobiotic biotransformation (e.g., drug metabolism) in the same individual. Such variation is not observed when alterations in P450 activity are determined genetically.
Due to their broad substrate specificity, it is possible that two or more P450 enzymes can contribute to the metabolism of a single compound. For example, two P450 enzymes, designated CYP2D6 and CYP2C19, both contribute significantly to the me-
tabolism of propranolol in humans: CYP2D6 oxidizes the aromatic ring to give 4-hydroxypropranolol, whereas CYP2C19 oxidizes the isopropanolamine side chain to give naphthoxylactic acid (see Fig. 6-23). Consequently, changes in either CYP2D6 or CYP2C19 do not markedly affect the disposition of propranolol. Three human P450 enzymes, CYP1A2, CYP2E1, and CYP3A4, can convert the commonly used analgesic, acetaminophen, to its hepatotoxic metabolite, N-acetylbenzoquinoneimine (Fig. 6-43). It is also possible for a single P450 enzyme to catalyze two or more metabolic pathways for the same drug. For example, CYP2D6 catalyzes both the O-demethylation and 5-hydroxylation (aromatic ring hydroxylation) of methoxyphenamine, and CYP3A4 catalyzes the 3-hydroxylation and N-oxygenation of quinidine, the M1-, M17-, and M21-oxidation of cyclosporine, the 1- and 4-hydroxylation of midazolam, the tert-butyl-hydroxylation and N-dealkylation of terfenadine, and several pathways of testosterone oxidation, including 1 -, 2 -, 6 -, and 15 -hydroxylation and dehydrogenation to 6-dehydrotestosterone (Figs. 6-36 and 6-43).
The pharmacologic or toxic effects of certain drugs are exaggerated in a significant percentage of the population due to a heritable deficiency in a P450 enzyme (Tucker, 1994; Meyer, 1994; Smith et al., 1998). The two major polymorphically expressed P450 enzymes are CYP2D6 and CYP2C19, although allelic variants have been described for nearly all of the human P450 enzymes involved in xenobiotic biotransformation (Smith et al., 1998). Individuals lacking CYP2D6 or CYP2C19 were initially identified as poor metabolizers of debrisoquine and S-mephenytoin, respectively. However, because each P450 enzyme has a broad substrate specificity, each genetic defect affects the metabolism of several drugs. The incidence of the poor-metabolizer phenotype varies among different ethnic groups. For example, 5 to 10 percent of Caucasians are poor metabolizers of debrisoquine (an antihypertensive drug metabolized by CYP2D6), whereas less than 1 percent of Japanese subjects are defective in CYP2D6 activity. In contrast, 20 percent of Japanese subjects are poor metabolizers of S-mephenytoin (an anticonvulsant metabolized by CYP2C19), whereas less than 5 percent of Caucasians are so affected. On Vanuatu and some other Pacific islands, as many as 70 percent of the population are CYP2C19 poor metabolizers (Kaneko et al., 1999). Some individuals have been identified as poor metabolizers of tolbutamide and phenytoin, both of which are metabolized by CYP2C9, or as poor metabolizers of coumarin or phenacetin, which are metabolized by CYP2A6 and CYP1A2, respectively. However, the incidence of each of these phenotypes is apparently less than 1 percent of the populations examined to date.
The observation that individuals who are genetically deficient in a particular P450 enzyme are poor metabolizers of one or more drugs illustrates a very important principle—namely that the rate of elimination of drugs can be largely determined by a single P450 enzyme. This observation seems to contradict the fact that P450 enzymes have broad and overlapping substrate specificities. The resolution to this apparent paradox lies in the fact that, although more than one human P450 enzyme can catalyze the biotransformation of a xenobiotic, they may do so with markedly different affinities. Consequently, xenobiotic biotransformation in vivo, where only low substrate concentrations are usually achieved, is often determined by the P450 enzyme with the highest affinity (lowest apparent Km) for the xenobiotic. For example, the N-demethylation of diazepam (shown in Fig. 6-40) and the 5-hydroxylation of omeprazole are both catalyzed by two human