

Quantum Chemistry of Solids / 22-LCAO Calculations of Perfect-crystal Properties
.pdf9.1 Theoretical Analysis of Chemical Bonding in Crystals |
357 |
The electronic structure for the MgO crystal was calculated in [608] both in the LCAO approximation and in the PW basis. In both cases the calculations were done by the density-functional theory (DFT) method in the local density approximation (LDA). The Monkhorst–Pack set of special points of BZ, which allows a convergence to be obtained (relating to extended special-points sets) in the calculations of electronic structure, was used in both cases. For the LCAO calculations the Durand–Barthelat pseudopotential [484] was used. In the case of the PW calculations the normconserving pseudopotential and a PW kinetic energy cuto of 300 eV were used.
For construction of the WTAOs the delta-functions were used as the weight functions in the variational functional (9.35). In this case, the variational procedure is equivalent to projecting the delta-functions onto the space of the states of chosen energy bands and demands a very low computational e ort.
In both types of calculations the s- and p-bands of oxygen atoms form the four valence bands. The s-band of the Mg in the case of the LCAO basis is the highest possible (twelfth) conduction band. For the PW calculations, the lower 32 conduction bands were examined. This analysis showed that the s-band of the Mg atom in PW calculations is also located among high-energy conduction bands. If one considers more than 32 conduction bands, the location of the s-band of the Mg atom can only increase, which would not a ect the results of the population analysis.
The calculations [608] showed that the WTAOs, corresponding to the LCAO and PW bases are essentially di erent in their form. This distinction is probably caused mainly by the di erence in the structure of the bases used for the LCAO and PW calculations. All the WTAOs, excluding the s-WTAO of the Mg atom in the PW calculations, are well localized. The poor localization of the latter is probably due to considering only 32 lower bands, while the actual s-band of the Mg atom might lie higher in the energy than the examined ones. Anyway, the di erence in the WTAOs’ form and localization level does not a ect the results of WTAO-based population analysis for MgO.
The atomic charges and covalences, calculated within the conventional approaches and WTAOs method, are given in Table 9.14.
Table 9.14. Calculated atomic charges and covalencies in MgO
Basis |
Population analysis |
Charge on Mg atom |
Atomic covalence |
||
|
|
|
Mg |
O |
|
LCAO |
Mulliken |
1.93 |
0.15 |
0.30 |
|
L¨owdin |
1.54 |
0.88 |
1.13 |
||
|
|||||
PW |
Mulliken |
1.06 |
1.63 |
1.65 |
|
L¨owdin |
0.90 |
1.90 |
1.89 |
||
|
|||||
LCAO |
Nonorthogonal WTAOs |
2.00 |
0.00 |
0.00 |
|
|
Orthogonal WTAOs |
1.97 |
0.05 |
0.05 |
|
PW |
Nonorthogonal WTAOs |
2.00 |
0.00 |
0.05 |
|
Orthogonal WTAOs |
1.93 |
0.13 |
0.14 |
||
|
|||||
The spilling parameter for the projection procedure (see Sect. 9.1.6) in the PW calculations was 2.5 × 10−3. It is seen from this table that among the traditional schemes, only the Mulliken analysis performed for the LCAO calculations exhibits a
358 9 LCAO Calculations of Perfect-crystal Properties
more or less ionic picture of chemical bonding in MgO. All the other conventional methods give the results corresponding to the mixed ionic-covalent type of bonding, which is actually quite unnatural for MgO. Besides, for the traditional methods the values of the atomic charges and covalences are significantly di erent depending on the calculation scheme and the type of basis.
On the contrary, the method based on orthogonal or nonorthogonal WTAOs correctly demonstrates the ionic nature of chemical bonding in MgO crystal whatever basis is used for the electronic-structure calculations. The values obtained within the di erent types of basis are close and correspond to the practically pure ionic character of bonding in this crystal. Though the LDA band-structure calculations for the MgO crystal in LCAO and PW bases give di erent values for the total energy and bandgap width and the forms of corresponding WTAOs are quite dissimilar, nevertheless, the results of population analysis performed in the basis of WTAOs are practically the same. This is a consequence of a high-energy location of the s-band of the Mg atom in both types of calculations.
9.1.5 The Localized Wannier Functions for Valence Bands: Chemical Bonding in Crystalline Oxides
The approach to the analysis of the chemical bonding in crystals discussed above is based on the population-analysis procedure, applied in the basis of AOs or WTAOs. In the second approach [63] the Wannier functions are generated using the Bloch functions of only the upper valence bands (see Chap. 3), so that the number NW F of WFs per primitive cell is equal to the number of valence bands under consideration. For metal oxides, considered in this section, the upper valence energy bands are formed by 2s and 2p states of oxygen atoms. Therefore, the number of WFs per cell NW F = 4Nf NO (Nf is the number of formula units per cell, NO is the number of oxygen atoms in the formula unit). In particular, NW F = 4, 24, 48 for MgO, Al2O3 and AlPO4 crystals with one, two and three formula units per cell, respectively. This approach provides the additional numerical characteristics of chemical bonding, which complement the population analysis in periodic systems.
Any of the WFs in the reference cell is given in the AO basis set (used in the Bloch-functions calculation) as
|
M |
|
|
|
|
Wi(r − g) = Wi(r) = |
g |
(9.44) |
Cµi χµ (r − Aµ − g ) |
µ=1 g
where the orthonormality condition Wig|Wig = δgg holds for the periodic images
Wi(r − g) of the reference cell WF. The coe cients Cg in (9.44) are related to the
µi
coe cients Cµi(kj ) in the Bloch functions of M valence bands by the relation
|
|
g |
(9.45) |
Cµi(kj ) = exp(−ikj g )Cµi |
g
In these relations µ = 1, 2, . . . , M numbers AOs in the primitive cell, g is the direct lattice translation vector, i = 1, 2, . . . , NW F numbers the valence bands and WFs and kj (j = 1, 2, . . . , L) are defined by the size of cyclic system L used for the summation
9.1 Theoretical Analysis of Chemical Bonding in Crystals |
359 |
over BZ (see Chap. 3). WFs are characterized in terms of their Mulliken atomic populations by
qg |
= |
|
Cg |
Cg+g Sg |
(9.46) |
A,i |
µ |
|
µ,i |
νi µν |
|
|
|
|
|
|
|
|
|
|
|
||
where the first sum runs over the AOs µ centered on atom A and the second one over all the AOs ν and cells g . The atomic populations (9.46) are normalized to one i.e., they satisfy the condition qA,ig = 1. These populations are used to describe
A,g
the localization properties of WFs, as qA,ig gives the fraction of the electron density of WF Wi(r) assigned to atom A in cell g. The range of WF Wi(r) localization is defined by nonzero values of (9.46) in spheres of atoms around the localization center of WF. The latter (centroid position) is defined by
r¯i = |
r |Wi(r)|2 dr = |
|
x¯sie¯s, x¯si = |
xs |Wi(r)|2 dr |
(9.47) |
s |
The centroid positions are used in the definition of the second-order central moment tensor, τi,lm of the electron density associated to WF Wi(r):
τi,lm = dr |Wi(r)|2 (rl − ri,l)(rm − ri,m) |
(9.48) |
where r = (r1, r2, r3) and ri = (ri1, ri2,ri3 ). Tensor (9.48) contains useful information on the shape and spatial extent of the WFs. Centroid position ri of the WF Wi(r)
also determines the so-called polarization fraction for Wi(r): |
|
|
||||||||||
|
|
(r |
| |
− |
| |
rA) |
|
|
|
|
|
|
Pi = |
2 |
|
i −rb |
rA |
−2 |
|
|
− 1 |
, (0 |
≤ Pi |
≤ 1) |
(9.49) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
which is proposed to estimate the degree of ionicity and covalency of the chemical bonding between atoms A and B (rA and rB are their position vectors). This parameter takes the value Pi = 1 for a pure ionic bond and Pi = 0 for a pure covalent bond. A and B atoms are supposed to give the two largest contributions of the Mulliken atomic population to the WF Wi(r).
These indices were used to analyze the chemical bonding in semiconductors [610] and metal oxides [63], based on HF LCAO calculations. In Table 9.15 the results for metal oxides are presented. In this table Nf , NW F and Nc are the number of formula units in the primitive cell, the number of valence-WFs in the primitive cell and the coordination number of the oxygen atom. According to the point symmetry at the O position, the four WFs can be equivalent, as in the case of MgO and MnO, or split into 2, 3, or 4 sets; the number of equivalent WFs in each set is indicated in the Neq column. In those systems where crossings occur between the valence and the most di use semicore bands (the transition-metal oxides MnO and ZnO and CaSO4) the cation d and the Ca 2p bands, respectively, have also been included within the active subspace. In MnO, where only the ferromagnetic spin polarized solution was considered, α and β subspaces are localized independently (the former conventionally containing the five Mn d states), [63]. In the right part of the table some features of the oxygen-WFs are provided: Neq is the number of equivalent oxygen-WFs in the set; q1, q2 and q3 are the three largest Mulliken atomic populations (the number of
360 9 LCAO Calculations of Perfect-crystal Properties
equivalent contributions is given in brackets!). The Or column indicates whether the second-order moment tensor principal axis is oriented along the bond axis (y) or not (n), and the last column classifies the WFs as covalent bond (cv), ionic (io), or lonepair (lp). The degree of localization of the oxygen WFs is estimated in terms of the
Table 9.15. Characterization of the oxides through their symmetry and WF features [63] (explanations are given in text)
System |
Space |
Nf |
NW F |
Nc |
Neq |
q1 |
q2 |
q3 |
Or |
Class |
group |
||||||||||
MgO |
F m3m |
1 |
4 |
6 |
4 |
0.991 |
0.005(2) |
0.004 |
n |
io |
MnO-α |
F m3m |
1 |
9 |
6 |
4 |
1.005 |
–0.001(4) |
— |
n |
io |
MnO-β |
— |
|
4 |
|
4 |
0.969 |
0.016(2) |
0.005 |
n |
io |
ZnO |
P 63mc |
2 |
18 |
4 |
1 |
0.929 |
0.065(3) |
0.002 |
y |
cv |
|
|
|
|
|
3 |
0.927 |
0.065(3) |
0.003 |
y |
cv |
Al2O3 |
R3c |
2 |
24 |
4 |
2 |
0.964 |
0.043 |
0.004 |
y |
cv |
|
|
|
|
|
2 |
0.942 |
0.060 |
0.003 |
y |
cv |
SiO2 |
P 3221 |
3 |
24 |
2 |
1 |
0.957 |
0.022 |
0.020 |
n |
lp |
|
|
|
|
|
1 |
0.947 |
0.029 |
0.026 |
n |
lp |
|
|
|
|
|
1 |
0.815 |
0.167 |
0.017 |
y |
cv |
|
|
|
|
|
1 |
0.812 |
0.171 |
0.017 |
y |
cv |
AlPO4 |
P 3121 |
3 |
48 |
2 |
1 |
0.965 |
0.030 |
0.006 |
n |
lp |
|
|
|
|
|
1 |
0.952 |
0.031 |
0.021 |
n |
lp |
|
|
|
|
|
1 |
0.910 |
0.070 |
0.023 |
y |
cv |
|
|
|
|
|
1 |
0.821 |
0.173 |
0.006 |
y |
cv |
CaSO4 |
P 6222 |
3 |
48 |
1 |
3 |
0.958 |
0.039 |
0.004 |
n |
lp |
|
|
|
|
|
1 |
0.711 |
0.296 |
0.001 |
y |
cv |
Mulliken atomic populations (9.46), attributed to oxygen and its neighboring atoms. These data are summarized in columns q1, q2, and q3 of Table 9.15. It turns out that, in all cases, most of the electron density is localized on the oxygen atom, q1 ranging from 0.71 (CaSO4) to 1.01 (MnO). The value of the second atomic contribution is never larger than 0.30 (CaSO4); in most cases, however, it is of the order of 0.05. The third largest atomic population is much smaller and never exceeds 0.03 (SiO2).
The Or column provides information as to whether the principal axis connected to the largest eigenvalue of the second-order moment tensor is oriented along the bond axis or not. The bond axis of an O-WF is defined as the line joining the oxygen and the atom that brings the second largest contribution to the total population. For ease of discussion, one can try to use the two kinds of information (qi and Or indices) to classify (with a certain degree of arbitrariness) the O-WFs bonds as covalent bond (cv), lone-pair (lp), or ionic (io). In particular, it is considered as cv, a WF oriented along the bond direction, (orientation criterion); alternatively, a WF can be classified as cv, when q1 < 0.95 and q2 > 0.05 (population criterion), where q1 and q2 are the two largest contributions to the total population (see Table 9.15). A non-cv-WF is classified as io or lp when the coordination number of O is larger or smaller than 4 (the number of WFs formally attributable to oxygen), respectively. As is shown in [63], io and lp WFs do have quite di erent characteristics. The two criteria provide a very
9.1 Theoretical Analysis of Chemical Bonding in Crystals |
361 |
consistent classification, the only exception being the most localized O-WFs of Al2O3, classified as cv or not according to the first or second criteria, respectively. Indeed in this case, both types of WFs are of the same chemical nature and quantitative differences are probably due to geometrical constraints. The orientation criterion seems to be the most appropriate and has been adopted (see last column of Table 9.15). This classification allows various trends to be traced with respect to the nature of the O-WFs along the series of compounds, as was done in [63]. In particular, it turns out that WFs give quite a complete picture of the chemical nature of the X–O bond, indicating that the covalent character increases according to the following sequence with respect to X: Mg < Mn < Al < Zn < Si < P < S. This order coincides with the cation Pauling electronegativities and demonstrates the atomic nature dependence of the chemical bonding in crystals.
This approach was applied also in [611] in the comparative study of chemical bonding in SrTiO3 and SrZrO3 crystals. The results of these calculations are discussed in the next section in the consideration of the projection technique for population analysis in crystals.
The numerical values of the indices dependence on the basis-set choice requires additional investigation. It is evident that use of L¨owdin atomic populations instead of Mulliken ones can change the localization of WFs, especially in the case when di use AOs are included in the basis set. From this point of view the WTAOs use is preferable. As was demonstrated above, the numerical values of local properties are close in Mulliken and L¨owdin population analysis made with WTAOs.
In the approach [63] WFs are generated directly from Bloch valence states without any preliminary symmetry analysis. In this case one obtains WFs that have the centroid positions in the vicinities of some points of the direct lattice of the crystal occupied by atoms or being midpoint between the pairs of symmetry-equivalent atoms (for the oxides listed in Table 9.15, these points are near oxygen atom positions). The noncontradictory chemical interpretation of the WFs obtained is di cult due to the absence of the symmetry analysis. Indeed, it is di cult to explain the appearance in MgO crystal (see Table 9.15) of four equivalent WFs corresponding to the tetrahedral sp3 hybridization, while the oxygen atom is octahedrally coordinated.
The improvements of this theory were suggested in [612]. The importance of the symmetry analysis in the procedure of the generation of maximally LOs in crystals was shown. It was suggested that the numerical parameters of the LOs corresponding to the valence energy bands of the crystal can be used in the theory of chemical bonding only if these functions form bases of irreducible representations of site-symmetry groups of atoms and bonds in crystals. The background of LOs symmetry analysis is the theory of band representations (BRs) of space groups, considered in Chap. 3. This analysis is applicable both to nonorthogonal atomic-like localized orbitals, centered on the atoms, and to localized Wannier orbitals (LWOs), i.e. to an orthonormal set of LOs. BRs of space groups are used to identify possible LOs symmetry from the symmetry of the canonical (Bloch) orbitals of the calculated energy bands or to establish the fact that the construction of LOs from the canonical orbitals chosen is impossible for the reasons of symmetry. In the general case, the LWOs are centered on some center q and transform according to some representation Re (in the general case reducible) of the site-symmetry group Lq of this center. Let us represent these
W (β) |
transforming according to IRs β |
functions as linear combinations of the LWOs !j |
362 9 LCAO Calculations of Perfect-crystal Properties
of the site group Lq, |
! |
|
|
(9.50) |
|
Wi(r) = |
αβj,iWj(β)(r), αT α = E |
β,j
The transformation, with the help of the matrix α, is orthogonal as it relates to two orthonormal bases in the space of the representation Re (E is the unit matrix). Inserting (9.50) in (9.47), one obtains
x¯si = |
Dβj,β(s) j αβj,iαβ j ,i |
(9.51) |
ββ jj |
|
|
|
|
(s) !(β) !(β )
where the number of independent values Dβj,β j = Wj (r)|xs|Wj (r) is equal to the number of irreducible components of the vector representation contained in the
symmetric square {Re × Re }+ of the representation Re [13]. If Wi(r) coincide with their irreducible components (αβj,i = δβj,i), then ri = 0 for centrosymmetric site
groups Lq as all Dβj,β(s) j = 0(¯xsi = 0). In the general case, as seen from (9.51), ri = 0 and their lengths and orientations depend on the value of the coe cients αβj,i. Being
functions of the centroid position, the polarization fractions Pi (9.49) also depend on the value of the coe cients αβj,i. It is obvious that in the general case one can not attribute to the centroid of a LWO some physical meaning. But a particular choice of LOs can attach to the centroid a common sense useful in the theory of chemical bonding.
LWOs WA(r) sited on atoms describe lone pairs or a purely ionic bonding (Pi = 1). LWOs WB (r) sited on interatomic bonds mean the presence of a covalent component of the chemical bonding (0 < Pi < 1), the degree of covalence being determined by the value of the polarization fraction Pi that takes the form
|
di |
− |
dA |
|
|
|
|
|
− |
|
|
||
Pi = 2 |
dB |
dA |
− 1 |
(9.52) |
||
where dA, dB , and di are positions of atoms |
A, B, and |
the centroid of the LWO Wi(r) |
||||
on the bond line A − B counted o some arbitrary point on it. In the latter case, the BR associated with the valence bands of the crystal has to include the BR induced from some IR β of the site-symmetry group Lqb of the bond. The value Pi = 0 corresponds to a pure covalent bond. Note that this BR is a composite one. Indeed, the group Lqb is a subgroup of the site group LqA of the atom A (and of the site group LqB of the atom B) lying on the bond. The IR β induces in the groups LqA some reducible representation that, in its turn, induces in the space group G a composite BR [13]. There is an exception when a middle point of the bond is a symmetry point. In this case, the IR of the site group of this point induces in the space group G a simple BR. This situation takes place, for example, in the Si crystal where the IR a1g of the site group D3d of the middle point of the bond Si–Si (Wycko position c) induces in the space group Oh7 of the crystal the BR with q-basis consisting of the canonical Bloch states of the four-sheeted upper valence band. This LWO describes the purely covalent bond (P = 0).
In [612] the following procedure was suggested to describe the chemical bonding in crystals by analyzing only occupied valence states in the basis of LOs.

9.1 Theoretical Analysis of Chemical Bonding in Crystals |
363 |
As the first step a symmetry analysis of valence band states is carried out. The purpose of this analysis is to find the q-basis indices of the band representation (BR) corresponding to the valence states of the crystal. These indices can consist of: (a1) the irreducible representations (IRs) of the site groups of the bonds (including those of symmetry points on the bond lines, if there are any); (a2) the IRs of site groups of atoms. If a composite BR has several variants to be represented as a sum of simpler ones, the preference has to be given to the BRs induced from site groups of bonds. This symmetry analysis allows the centroid positions (on atoms or on bonds) of the LOs, generated from the Bloch functions of the valence states, to be found. The numerical characteristics of only these symmetry-defined LOs are suggested to give a noncontradictory unambiguous interpretation of chemical bonding in crystals. It was proposed in [612] to introduce not only the centroids position, but also the other numerical characteristics of the chemical bond related to the centroids position:
1) Contributions Q(iA) and Q(iB) to charges on atoms A and B related to the LO Wi(r)
(A) di − dB
Qi = 2 dB − dA ,
(B) dA − di
Qi = 2 dB − dA ,
−2 ≤ Qi(A), Qi(B) ≤ 0 |
(9.53) |
An electron charge Q(iA) + Qi(B) = −2 associated with the fully occupied LO is partitioned between atoms A and B and is inversely proportional to the distances between the centroid position and the atoms.
2) The degree of ionicity Ii of the bond A − B related to the LO Wi(r),
|
|
|
I |
= |
|
2di − dA − dB |
|
= |
1 |
|
Q(A) |
− |
Q(B) |
|
, |
|
0 |
≤ |
I |
i ≤ |
1 |
|
(9.54) |
||||||||||
|
|
|
|
2 |
|
|
|||||||||||||||||||||||||||
|
|
|
i |
|
|
|
dB − dA |
|
|
|
|
|
i |
i |
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The degree of covalency |
Ki of the bond |
A–B related to the LO Wi(r), |
|
||||||||||||||||||||||||||||||
K |
|
= 1 |
|
|
2di − dA − dB |
|
= 1 |
|
1 |
|
|
Q(A) |
|
Q(B) |
|
, |
0 |
|
|
K |
|
|
1 |
(9.55) |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||
i |
− |
|
− 2 |
|
≤ |
i |
≤ |
||||||||||||||||||||||||||
|
|
|
|
dB |
− dA |
|
|
|
i |
|
− i |
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ionicity Ii |
coincides |
with the polarization |
fraction (9.52). |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
For pure ionic bonding Q(iA) = −2, Q(iB) = 0 (or vice versa), K = 0, I = 1. For pure covalent bonding Q(iA) = Q(iB) = −1, K = 1, I = 0. The definitions (9.52)–(9.55) are introduced in such a way that the LWO centroid position defines the characteristics of chemical bonding. In fact, the details of the electron density distribution have to be included, as is done in the definition (9.48) of the second-order central moment tensor.
In the second step of the chemical-bonding analysis the space Ω of the states of all the valence energy bands is projected on the spaces Ωi of IR of the site-symmetry groups related to the components of the BR in the q-basis established above.
The third step is the generation from the canonical Bloch valence states maximally localized orbitals in the spaces Ωi by any known procedure.
The fourth step consists of calculation of numerical characteristics of the chemical bonding on the base of these LOs. This calculation includes both the characteristics, defined by the centroids positions, and those depending on the details of the electron density distribution.
364 9 LCAO Calculations of Perfect-crystal Properties
As follows from the theory of BR, the LOs obtained without any preliminary symmetry analysis, are in fact to be bases of some representations (in the general case reducible) of site-symmetry groups of the Wycko positions of atoms and bonds, the preference has to be given also to BRs induced from site groups of bonds. In this case, steps 2 and 3 have to be replaced by: (2a) Generation maximally LOs in the space of the canonical Bloch valence states by any known procedure. (3a) A proper symmetrization of generated LOs according to the results of the symmetry analysis of step 1.
We shall illustrate the discussed procedure using the example of the SrZrO3 crystal with the cubic perovskite structure. In Table 9.16 band representations of space group Oh1 induced by those atomic states that take part in the valence band formation are given. Each line of this table defines the connection of the q-basis for site-symmetry groups Oh(Sr), D4h(O) with the k-basis at the symmetry points of BZ, see also Sect. 3.3.
Table 9.16. Band representations of upper valence bands in the SrZrO3 crystal induced from Sr 4p-, O 2s- and O 2p-atomlike states
Atom |
q-basis |
Γ |
R |
M |
X |
|
states |
||||||
|
|
|
|
|
||
Sr 4p |
(b, t1u) |
4− |
5+ |
2−5− |
1+5+ |
|
O 2s |
(d, a1g ) |
1+3+ |
4− |
1+5− 1+2+3− |
||
O 2pz |
(d, a2u) |
4− 1+3+ 1+2+3− 1+5− |
||||
O 2px,y |
(d, eu) |
4−5− 4+5+ 3+4+5± 3−4−5± |
||||
To include the electron-correlation e ects on the chemical bonding, the calculations of SrZrO3 crystal [612,613] were performed by the DFT LCAO method with the CRYSTAL03 code [23] and the PBE exchange-correlation potential used in [614] for the geometry optimization in the generalized gradient approximation (GGA). For the Sr and Zr atoms, the Hay–Wadt pseudopotentials [483] in the small-core approximation were used. In this case, the 4s, 4p, and 5s states of the Sr atom in the 4s24p65s2 configuration and the 4s, 4p, 4d, and 5s states of the Zr atom in the 4s24p64d25s2 configuration were treated as valence states. The basis functions were taken as the atomic functions of the Sr atom from [606] and the 8-411G * functions obtained for the Zr atom in [615] with optimization of the 5sp, 6sp, and 5d outer orbitals for the ZrO2 crystal with experimental geometric parameters. For the oxygen atom, the fullelectron basis set [606] describing the oxygen atom in the 1s22s22p4 configuration was used. The Monkhorst–Pack 8×8×8 set of special points was used in the BZ sampling. The lower valence bands are composed of Sr 4p- and O 2s-states (six sheets), whereas the upper valence band is produced mainly by the O 2p-atomic orbitals (nine sheets), see Fig. 9.2. The symmetry of all these valence-band states and the atomic nature of the projected density of states is the same both in these DFT-LCAO and in former DFT-PW calculations [616]. The iterative procedure implemented in CRYSTAL03 code was applied to generate LOs using crystal orbitals of these 15 occupied bands. The Bloch functions for the Γ point of the BZ are taken as the starting LOs. As a result, the iterative procedure gives 15 LOs. Three of them are centered on the Sr
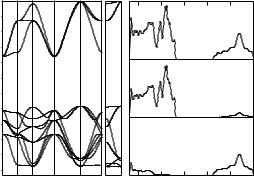
9.1 Theoretical Analysis of Chemical Bonding in Crystals |
365 |
|
4.0 |
a |
|
|
|
|
|
|
|
|
3..0 |
|
|
|
|
2.0 |
|
|
|
eV |
1.0 |
|
|
|
0.0 |
|
|
|
|
Energy, |
|
|
|
|
-1.0 |
|
|
|
|
-2.0 |
|
|
|
|
-3.0 |
|
|
|
|
|
|
|
|
|
|
-4.0 |
|
|
|
|
-5.0 |
|
|
|
|
-6.0 |
|
|
|
|
-7.0 |
|
|
|
|
|
X M |
|
R X M R |
|
|
|
k |
|
|
b |
|
|
|
|
|
|
Total |
|
|
|
|
|
O: p |
|
|
|
|
|
Zr:d |
|
|
|
-6.0 |
-4.0 |
-2.0 |
0.0 |
2.0 |
4.0 |
Energy, eV
Fig. 9.2. Electronic structure of SrZrO3 crystal: (a) band structure; (b) total and projected DOS. LCAO DFT calculations [613]
atom and are connected with Sr atom 4p-states. The remaining 12 LOs are connected with the three symmetry-equivalent oxygen atoms in the crystal, four LOs with the centroid positions near each oxygen atom. To analyze the nature of chemical bonding with these LOs, the information about the symmetry of the band states induced from Sr 4p-, O 2s-, and O 2p-states forming two upper valence bands given in Table 9.16 is used. It is seen that these states form a basis of composite BR described in q-basis by the symbol
(b, t1u) + (d, a1g + a2u + eu) |
(9.56) |
Therefore, four LWOs Wi(r) with the centroid positions near each oxygen atom form a basis of a four-dimensional representation a1g + a2u + eu of the site group D4h of the oxygen atom. Let us denote LOs that are the basis functions of these IRs as
4 |
(! |
! |
! |
|
! |
! |
! |
! |
|
follows: W1 |
(r) = W1(d,eu ) |
(r), W2 |
(r) = W2(d,eu ) |
(r), W3 |
(r) = W3(d,a2u ) |
(r), W4 |
(r) = |
||
W (d,a1g ) |
r). Then, instead of (9.50), one has |
|
|
|
|
||||
! |
|
|
|
4 |
! |
|
|
|
|
|
|
|
j |
i = 1, 2, 3, 4 |
|
(9.57) |
|||
|
|
Wi(r) = |
|
αjiWj (r), |
|
||||
|
|
|
|
=1 |
|
|
|
|
|
The starting LWOs of the computer code [23] result in Wi(r) (9.57) with some definite values of the coe cients αji. In this case, Re = eu + a2u + a1g , the representation {Re × Re }(+) = 3a1g +b1g +b2g +eg +eu +a2u contains only one times two irreducible components of the vector representation (eu +a2u), i.e. there are only two independent parameters in (9.51),
3
r¯i = x¯sie¯s, x¯si = Dsα4iαsi |
(9.58) |
s=1 |
|
where

366 |
|
9 LCAO Calculations of Perfect-crystal Properties |
|
|
= 2 !4 |
|
3|!3 |
|
|
|||||||
D |
1 |
!4 |
| 1|!1 |
|
= D |
2 |
!4 |
| 2|!2 |
|
, D |
3 |
| |
(r) |
|
||
|
= 2 W |
(r) x W |
(r) |
|
= 2 W |
(r) x W |
(r) |
|
W |
(r) x |
W |
|
||||
(9.59) In Table 9.17 the Cartesian coordinates x¯si of centroid positions and the polarization fractions Pi for LWOs Wi(r) evaluated by the computer code [23] are given. It is evident (see (9.58)) that the other choice of the starting LWOs can, in principle, give other centroid positions and other values of Pi. Let us note that the BR (d, a1g +a2u) is
Table 9.17. Centroids positions (x1i, x2i, x3i), in a.u., and polarization fractions pi for functions Wi(r) in the SrZrO3 crystal localized according to the Wannier–Boys mixed scheme [63]
Wi x1i |
x2i |
x3i |
pi |
W1 –0.039 0.465 –0.002 0.999
W2 0.465 –0.039 –0.002 0.999
W3 –0.211 –0.211 0.774 0.610
W4 –0.215 –0.215 –0.769 0.612
a composite one. The points on the line binding Zr and O atoms (Wycko position e) have the site-symmetry C4v . The IR a1 of this group induces in the group D4h (the site symmetry group of the oxygen atom) the representation a1g + a2u : a1(C4v ) ↑ D4h = a1g + a2u(D4h). Therefore, instead of (9.56) one can write another symbol in q-basis
(b, t1u) + (d, eu) + (e, a1) |
(9.60) |
for the same BR corresponding to the 15-sheeted band chosen. Note that the IR a1 of C4v induces also the representation a1g + eg + t1u of the site group Oh of the Zr atom – the second atom on the bond line Zr–O: a1(C4v ) ↑ Oh = a1g + eu + t1u(Oh). Thus, one can attribute one more symbol in the q-basis to the BR corresponding to the same 15-sheeted energy band,
(b, t1u) + (d, eu) + (a, a1g + eg + t1u) |
(9.61) |
It is seen that the part (b, t1u) + (d, eu) is common for three symbols (9.56)–(9.60)– (9.61) of the BR under consideration. They correspond to LWOs, localized on Sr and oxygen atoms. The rest of the BR is given in three di erent ways by its labels (9.56)- (9.60)-(9.61). The symmetry considerations show that it is possible to generate from the states of valence bands of the SrZrO3 crystal the LWOs centered on zirconium and on oxygen atoms. These LWOs form the bases of reducible representations of the site groups of Zr and O atoms, a1g + eg + t1u of Oh and a1g + a2u of D4h, respectively. Linear combinations of functions of any of these bases are equivalent from the grouptheoretical point of view, but will give di erent values for the numerical characteristics of chemical bonding (9.52)–(9.55). However, there is only one possibility to reconcide these two bases. Both bases are induced from the same IR a1 of the common subgroup C4v (the site group of the Wycko position e on the bond line Zr–O) of the groups
Oh and |
D4h. |
|
|
|
|
The |
BR with q-basis (9.60) corresponds to |
a particular |
choice of |
the |
linear |
|
W |
transformed |
according |
to |
the IR |
combinations of !j (r) (9.57): two of them are |
|||||