2) Квадрупольный
Ионный пучок направляют в пространство между четырьмя параллельными электродами. Под действием электрических полей заряженные частицы колеблются, и при фиксированных значениях частоты и амплитуды поля ионы с определенным значением m/z проходят через анализатор. Остальные частицы сталкиваются и выбиваются из потока. Чтобы «пропустить» ионы с другим значением m/z, меняют либо частоту, либо амплитуду
«+»:
-
высокая чувствительность
-
небольшие размеры
-
невысокая цены
-
быстрая регистрация спектра
«-»:
-
верхний предел пропускания находится между m/z 1000 и 2000
3) Трехмерная ионная ловушка
Система электродов создает поле, позволяющее удерживать ионы достаточно долгое время (в зависимости от их массы). Импульсное изменение амплитуды напряжения на центральном электроде заставляет ионы с определенным m/zпереходить на нестабильные траектории и покидать ловушку, попадая в систему регистрации.
4) Времяпролетный
Его действие основано на зависимости скорости движения ионов от их массы. Их особенность – ионы движутся в бесполевом пространстве.
После ускорителя все ионы обладают одинаковой кинетической энергией, следовательно, чем больше их масса, тем меньше скорость, тем больше время пролета иона через анализатор.
4: Детекторы
1) применяют диодные вторично-электронные умножители, в которых ион, попадая на первый диод, выбивает из него пучок электронов, которые в свою очередь, попадая на следующий диод, выбивают из него еще большее количество электронов
2) микроканальные умножители, системы типа диодных матриц и коллекторы, собирающие все ионы, попавшие в данную точку пространства (коллекторы Фарадея)
Интерпретация масс-спектра:

8. ЯМР-спектроскопия. Общие физические основы, устройство спектрометра. Интерпретация одномерного ЯМР-спектра, связь строения молекулы с видом спектра. Спектроскопия на ядрах углерода, азота, фосфора: отличия, особенности получаемой информации.Импульсная ЯМР-спектроскопия, двумерные спектры: решение структурных задач методами ЯМР.
Спектроскопия ядерного магнитного резонанса – вид спектроскопии, которая регистрирует переходы между магнитными энергетическими уровнями атомных ядер, вызываемые радиочастотным излучением (резонансное поглощение или излучение электромагнитной энергии веществом, содержащим ядра с ненулевым спином во внешнем магнитном поле, на частоте ν (называемой частотой ЯМР), обусловленное переориентацией магнитных моментов ядер). (не разрушающий метод)
Только ядра со спиновым квантовым числом I, отличным от «0», могут вызывать сигнал ЯМР, или быть активными в ЯМР (Сигналы в спектрах ЯМР могут давать только ядра атомов, обладающих нечетным спиновым числом. Таким образом, наиболее распространенные изотопы углерода 12С, кислорода 16О и многие другие, например, дейтерий, являясь немагнитными, не регистрируются в ЯМР-спектрах. Из ядер атомов, наиболее часто встречающихся в органических соединениях, магнитным моментом обладают изотопы 1Н, 13С, 19F, 31P, 15N, 17O)
В сущности, эксперимент ЯМР состоит в том, чтобы сообщить энергию ядру и перевести его с одного энергетического уровня на другой, более высокий уровень. Поскольку точное значение ΔЕ (разность энергий двух соседних уровней) зависит от молекулярного окружения возбуждаемого ядра, имеется возможность связать величину ΔЕ со строением молекулы и в конечном итоге определить структуру всей молекулы.
Физические основы:большинство ядер обладает угловым моментом количества движения Р

ħ – постоянная Планка
I– квантовое число углового момента (ядерный спин)
Магнитный момент (наличие у ядра углового момента приводит к наличию у ядра магнитного момента): μ = γР
γ – гиромагнитное отношение ядра (const для каждого ядра)

Магнитное поле, в котором находится ядро, зависит от структуры в-ва
z, B0
N,
αM0m
= +1/2













N,βm
= -1/2
Протоны в поле 𝐁0 распределяются на двух уровнях –– одни спины выстраиваются по полю 𝐁0 (α-спины, α-состояние), другие –– против (β-спины, β-состояние). Ориентация сопровождается выделением или поглощением кванта энергии.

ОСНОВНОЕ УРАВНЕНИЕ ЯМР: ν = γН0, где
ν – резонансная частота ядра;
γ – гиромагнитное отношение ядра (const для каждого ядра);
Н0 – напряженность внешнего магнитного поля.
Основные характеристики спектра ЯМР:
-
количество сигналов = количество неэквивалентных ядер данного типа
-
положение сигналов (химический сдвиг) = распределение электронной плотности по молекуле
-
форма сигналов (спиновое расщепление) = тип и количество соседних ядер, конформация
-
площадь сигналов = количество эквивалентных ядер, давших сигнал

Химический сдвиг – степень ослабления поля (уменьшение резонансной частоты) относительно какого-либо стандарта (если протоны одной молекулы имеют одинаковый химический сдвиг и химически равноценны, то они называются химически эквивалентными)
В этаноле в метильной группе на атомах водорода высокая плотность, поэтому эта группа в сильном поле. В гидроксильной группе кислород стягивает на себя электронную плотность, поэтому в слабом поле.
δ = (ν – νэталон)/ν0 (измеряется в ppm – миллионные доли)
Спин-спиновое взаимодействие
Магнитное ядро «чувствует», сколько других магнитных ядер находится в молекуле рядом с ним в его ближайшем окружении.
Рассмотрим (в этаноле), как протоны группы СН2 могут оказывать влияние на экранирование протонов метильной группы. Каждый из двух этих протонов имеет свой магнитный момент, т.е. его можно рассматривать как микроскопический магнитик. Очевидно, что ориентации двух таких «магнитиков» в группе СН2 могут быть представлены тремя комбинациями. Магнитные моменты обоих протонов в одних молекулах могут быть ориентированы «по полю В0», а в других - «против поля В0». Вероятность встретить третью комбинацию «один – по полю, другой – против поля» будет (по статистике) в два раза больше, чем две первых. В тех молекулах, где оба «магнитика» протонов группы СН2 ориентированы «по полю В0», создаваемое ими дополнительное поле В' будет складываться с В0. В результате на протоны метильной группы будет действовать эффективное поле (В0+ В'), и резонансный сигнал группы СН3 таких молекул при развертке спектра появится в более слабом поле. Напротив, в молекулах, где оба «магнитика» протонов группы СН2 ориентированы «против В0» создаваемое ими дополнительное поле В' будет вычитаться из В0. Резонансный сигнал группы СН3 таких молекул появится в более сильном поле. Если же имеет место третья комбинация, то влияния двух маленьких «магнитиков» компенсируют друг друга. Поэтому центральная самая интенсивная компонента триплета будет расположена точно там, где она должна находиться при отсутствии влияния соседних протонов. Аналогично можно рассмотреть влияние трех протонов метильной группы на экранирование протонов метиленовой группы. Нетрудно подсчитать, что сигнал двух протонов метиленовой группы должен представлять собой квадруплет с соотношением интенсивности его компонентов 1:3:3:1.

Эквивалентность атомов:
-
химически-эквивалентные (одинаковое расстояние до соседей):
- гомотопные (переводятся друг в друга вращением)
- энантиотопные (переводятся друг в друга отражением)
-
магнитно-эквивалентные (одинаковые константы спин-спинового взаимодействия с соседями)
Спектр 1 порядка: расстояние между любыми двумя сигналами отличается более, чем в 10 раз
-
Спектр смеси соединений представляет собой суперпозицию спектров каждого индивидуального компонента смеси с отношением интенсивностей сигналов, равным мольным долям компонентов, разделенным на количество протонов в функциональной группе
-
Спин-спиновое взаимодействие между магнитно-эквивалентными ядрами не проявляются в спектре
-
Для ядер со спиновым квантовым числом I = ½ мультиплетность сигнала равна n+1, где n – число ядер в соседней группе
-
Расстояния между линиями мультиплетов в Гц соответствуют константам спин-спинового взаимодействия между рассматриваемыми ядрами
-
Относительные интенсивности линий внутри мультиплета соответствуют коэффициентам биномиального ряда. Эти коэффициенты можно определить из треугольника Паскаля
-
Величина спин-спинового взаимодействия в общем уменьшается при возрастании числа связей, разделяющих взаимодействующие ядра
Протоны, входящие в состав быстро обменивающихся групп (ОН, NH2) не дают обычной картиныспин-спинового взаимодействия, не наблюдается и расщепление сигналов соседних групп
Сложные спиновые системы не описываются приближением первого порядка.
Признаками отклонения от приближения 1 порядка является «неправильное» распределение интенсивностей линий в мультиплете, появление дополнительных линий
Разность заселенностей верхнего и нижнего уровней для ядра в магнитном поле, между которыми осуществляется переход, ничтожно мала. Интенсивность сигнала ЯМР определяется количеством поглощенной энергии, а ее поглощение прекращается сразу же после того, как заселенности уровней выравниваются (наступает «насыщение»). Именно поэтому для ЯМР исключительно важное значение приобретают процессы релаксации, которые возвращают систему к равновесному состоянию. Для того чтобы вернуться на нижний энергетический уровень, магнитное ядро должно потерять квант энергии, которое оно поглотило.
ЯМР СПЕКТРОМЕТР:

Требования:
-
магнит должен давать однородное поле высокой напряженности
-
разрешающая способность (отношение минимальной разности частот двух спектральных линий, которые прибор еще «видит как отдельные», к его рабочей частоте)
«Накопление» спектра – процесс сложения n спектров
Простейший эксперимент ЯМР:
Импульс
Накопление FT
спектр



PW, мксD0, мксAQ, с RD, с (задержка для полной релаксации)
Естественное разрешение (при условии, что релаксация не наступает раньше AQ) = 1/AQ
Спектроскопия на ядрах углерода, водорода и фосфора:наиболее часто в исследованиях используются ядра 1Н, 2Н, 13С, 31Р
2Н – анализ гидрофобной части липидного бислоя
31Р – анализ гидрофильной части липидного бислоя
Все три изотопа водорода (протий, дейтерий, тритий) имеют ядра, обладающие магнитными свойствами, у ядер других изотопов магнитных свойств может и не быть. К ним относятся, например, ядра углерода С и кислорода О. Отсутствие магнетизма у этих ядер не является недостатком ЯМР-спектроскопии, анаоборот, ее преимуществом. Если бы основные изотопыназванных элементов имели ядра с магнитными свойствами, многие спектры органических молекул, содержащих углерод и кислород, были бы сложнее, чем это наблюдается в действительности. Для исследования методом ЯМР именно ядер углерода можно воспользоваться другим изотопом этого элемента — 13С, ядра которого обладают магнитными свойствами и, несмотря на низкое естественное содержание (около 1%), дают возможность получить спектр ЯМР.
Импульсная ЯМР-спектроскопия: для увеличения соотношения сигнал-шум в ЯМР-спектрометрах пробу облучают кратковременными импульсами радиочастотного излучения. Длительность импульса составляет порядка 10 мкс, а промежуток между импульсами Т – от 1 до 10 с. В течение времени Т измеряют затухающий сигнал в направлении, перпендикулярном приложенному магнитному полю. Для увеличения отношения сигнал-шум регистрацию спектра повторяют многократно, а полученные сигналы суммируют (накапливают). При этом получают спектр ЯМР во временном представлении. Чтобы превратить полученные данные в обычный спектр ЯМР (в частотном представлении), используют преобразование Фурье.
Двухмерная ядерная магнитно-резонансная спектроскопия — один из видов ядерной магнитно-резонансной спектроскопии, в котором данные распределены в пространстве по двум осям. Виды двухмерной ЯМР: корреляционную спектроскопию (COSY), J-спектроскопию, обменную спектроскопию (EXSY), ядерную спектроскопию с эффектом Оверхаузера (NOESY). Двухмерная ЯМР удобна в установлении структуры сложных молекул, структуру которых тяжело установить с помощью одномерной ЯМР.










9. Хроматография: общие принципы, роль в решении задач биотехнологии. Сорбенты и элюенты, разделение веществ по их физико-химическим характеристикам. Жидкостная хроматография на прямой и обратной фазе, тонкослойная хроматография, гель-проникающаяи ионообменная хроматография. Газо-жидкостная хроматография, GCMS, LCMS.
Хроматография - физико-химический метод разделения и анализа смесей, основанный на распределении их компонентов между двумя фазами – неподвижной (сорбент) и подвижной (элюент), протекающей через неподвижную. Хроматографический анализ является критерием однородности вещества: если каким-либо хроматографическим способом анализируемое вещество не разделилось, то его считают однородным (без примесей).
Характерная особенность – многократность повторения процессов адсорбции-десорбции, ионного обмена или распределения между фазами.
Принципиальным отличием хроматографических методов от других физико-химических методов анализа является возможность разделения близких по свойствам веществ. После разделения компоненты анализируемой смеси можно идентифицировать (установить природу) и количественно определять (массу, концентрацию) любыми химическими, физическими и физико-химическими методами.
В некоторых случаях для идентификации веществ используется хроматография в сочетании с другими физико-химическими и физическими методами, например с масс-спектрометрией, ИК-, УФ-спектроскопией и др.
Основные достоинства хроматографического анализа:
-
экспрессность; высокая эффективность; возможность автоматизации и получение объективной информации;
-
сочетание с другими физико-химическими методами;
-
широкий интервал концентраций соединений;
-
возможность изучения физико-химических свойств соединений;
-
осуществление проведения качественного и количественного анализа;
-
применение для контроля и автоматического регулирования технологических процессов.
Сорбенты: полярные (силикагель, активированная окись алюминия, цеолиты), неполярные (активированный уголь, тефлон, сажа).
Элюенты: бензиновые фракции, циклогексан, трихлорэтан, толуол, дихлорметан, хлороформ, диэтиловый эфир, этилацетат, ацетон, этанол, вода
Требования к подвижной фазе:
- химическая инертность к компонентам смеси
- малая вязкость
- нелетучесть при температуре колонки
- химическая термостойкость
- высокая селективность
- хорошая растворяющая способность
A
tR1
tR2



tm

t
tR’ = tR - tm
Удерживаемый объем VR = F*tR
F – объемная скорость потока подвижной фазы
tm – мертвое время, время выхода элюента
Фактор удерживания (емкости): (оптимум 1,5<k<4)

Фактор разделения (коэффициент селективности): (оптимум α >1)
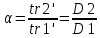
Эффективность колонки:высота слоя, эквивалентная теоретической тарелке (Н)

L – длина колонки
n– число теоретических колонок

Разрешение колонки:зависит от α,k,n (разрешение полное при Rs>1,5)

Размывание пиков:
-
вихревая диффузия (является следствием беспорядочного изменения линейной скорости около среднего значения, одни молекулы движутся быстрее, передвигаясь по свободным каналам между частицами наполнителя, другие — медленнее, проникая в замкнутые каналы)
-
молекулярная диффузия(обусловлена миграцией молекул, главным образом, в подвижной фазе и участков полосы с большей концентрацией в направлении, где концентрация меньше)
-
малая скорость процессов сорбции-десорбции
Жидкостная хроматографияэто метод разделения и анализа сложных смесей веществ, в котором подвижной фазой служит жидкость. Он применим для разделения более широкого круга веществ, чем метод газовой хроматографии. Это связано с тем, что большинство веществ не обладает летучестью, многие из них неустойчивы при высоких температурах (особенно высокомолекулярные соединения) и разлагаются при переводе в газообразное состояние. Разделение веществ жидкостной хроматографией чаще всего производится при комнатной температуре.
НормальнофазоваяЖХ – распределительная хроматография, в которой используется полярная неподвижная фаза, сильно удерживаются полярные компоненты (лучше разделяются)
Обращеннофазовая ЖХ – распределительная хроматография, в которой используется неполярная неподвижная фаза, сильно удерживаются неполярные компоненты (лучше разделяются)
Изократическое разделение – полярность полярной фазы не меняется
Градиентное разделение – полярность полярной фазы изменяется в ходе разделения
Детекторы:
-
оптические – УФ, рефракция, флуоресценция
-
масс-спектрометрические
-
электрохимические
Количественное определение:
-
Метод нормировки (Площадь пика вещества/сумма площадей = % вещества в смеси)
-
Метод калибровки (снимают хроматограммы для каждого компонента, строят график зависимость площади пика от количества введенного вещества)
-
Метод метки (сравнивают площади и высоту пика стандарта с исследуемым)
Ионообменная хроматография– метод разделения и анализа веществ, основанный на эквивалентном обмене ионов анализируемой смеси и ионообменника (ионита). Происходит обмен ионами между фазами гетерогенной системы. Неподвижной фазой являются иониты; подвижной, как правило, вода, т.к. этот элюент обладает хорошими растворяющими и ионизирующими свойствами.
Иониты – полимеры природного и синтетического, органического и минерального происхождения, содержащие ионогенные группы. Иониты имеют разветвленную матричную структуру, в состав которой входят фиксированные ионы. В зависимости от заряда иона матрица имеет положительный или отрицательный заряд, который компенсируется подвижными противоионами.
Гель-проникающая (эксклюзионная)– разновидность хроматографии, в ходе которой молекулы веществ разделяются по размеру за счёт их разной способности проникать в поры неподвижной фазы. При этом первыми выходят из колонки наиболее крупные молекулы (большей молекулярной массы), способные проникать в минимальное число пор стационарной фазы.
Последними выходят вещества с малыми размерами молекул, свободно проникающие в поры. В отличие от адсорбционной хроматографии, при гель-фильтрации стационарная фаза остаётся химически инертной и с разделяемыми веществами не взаимодействует.
Аффинная хроматография – разновидность лигандной хроматографии. В основе лежит реакция взаимодействия разделяемых примесей с лигандом, связанным с инертным носителем.
В случае аффинной хроматографии в роли примесей выступают биологически активные вещества (белки, ферменты), вступающие с лигандом (тоже, как правило, органическим) в специфическое биохимическое взаимодействие. Например: антитело-антиген, гормон-рецептор и т. д. Именно высокая специфичность подобного взаимодействия обусловливает высокую эффективность аффинной хроматографии и её широкое распространение.
ТСХ(не является количественным методом)- хроматографический метод, основанный на использовании тонкого слоя адсорбентав качестве неподвижной фазы. Он основан на том, что разделяемые вещества по-разному распределяются между сорбирующим слоем и протекающим через него элюентом, вследствие чего расстояние, на которое эти вещества смещаются по слою за одно и то же время, различается.
Размер пятна зависит от свойств вещества

В


А
Способы проявления пятен: УФ, реагенты-проявители (нингидрин, перманганат, ванилин, молибденовый синий)
ВЭЖХ: Отличительной особенностью ВЭЖХ является использование высокого давления (до 400 бар) и мелкозернистых сорбентов (обычно 3—5 мкм, сейчас до 1,8 мкм). Это позволяет разделять сложные смеси веществ быстро и полно (среднее время анализа от 3 до 30 мин)
Насос – инжектор – колонка – детектор – компьютер
Газовая хроматография–подвижной фазой является инертный газ (газ-носитель): газо-твердофазная (газо-адсорбционная), газо-жидкостная.
Газо-жидкостная хроматография — разделение газовой смеси вследствие различной растворимости компонентов пробы в жидкости или различной стабильности образующихся комплексов. Неподвижной фазой служит жидкость, нанесенная на инертный носитель, подвижной — газ.
Этот
метод можно использовать для анализа
газообразных, жидких и твёрдых веществ
с молекулярной массой меньше 400, которые
должны удовлетворять определённым
требованиям, главные из которых —
летучесть,
термостабильность, инертность, лёгкость
получения.

GCMS (ГХ+МС):в ходе хроматографирования регистрируют во времени интенсивность какого-либо пика с определенным массовым числом. В результате получается зависимость сигнала детектора от времени, как в хроматографии
LCMS (ВЭЖХ+МС):метод предназначен для анализа смесей труднолетучих, полярных веществ, не поддающихся анализу методом ГЖХ. В этом случае часть жидкого потока пропускают через капилляр (диаметр несколько микрон), в результате чего образуются капли, которые далее попадают в обогреваемую зону, где большая часть растворителя испаряется, а оставшаяся вместе с веществом попадает в ионный источник и ионизируется.
Возможности LCMS:
-
автоматический и ручной ввод образца
-
диапазон измеряемых масс от 20 до 20000 а.е.м. и выше
-
программируемое изменение температуры хроматографической колонки до кипения растворителя
-
анализ проб в растворе
-
возможность анализа полимеров, олигомеров, биологических объектов, полярных соединений
-
возможность подобрать колонку под узкоспециализированные задачи (нефтепродукты, ароматические соединения, лекарственные препараты)
10. Методы исследования молекулярных ансамблей и компонентов живых систем - клеточных органелл и др. Рентгеноструктурный анализ и дифракционные методы: принципы и применение в БТ и смежных областях. Достоинства и недостатки РСА, современные направления развития.
Методы исследования молекулярных ансамблей и компонентов живых систем:
-
Световая микроскопия (недостаточно разрешения для изучения органелл)
-
Электронная микроскопия (возможно наблюдать органеллы)
-
Люминесцентная и фазово-контрастная микроскопия (наблюдение хлорофилла, наблюдение в объеме)
-
Гистохимические методы (окраска)
-
Метод микрохирургии (изучение ядра)
-
Рентгеноструктурный анализ и другие дифракционные методы
Рентгеноструктурный анализ – метод исследования структуры вещества по распределению в пространстве и интенсивностям рассеянного на анализируемом объекте рентгеновского излучения
Рентгеновские лучи – электромагнитное ионизирующее излучение, занимающее спектральную область между гамма и ультрафиолетовым излучением в пределах длин волн от 10-12 до 10-5 см.
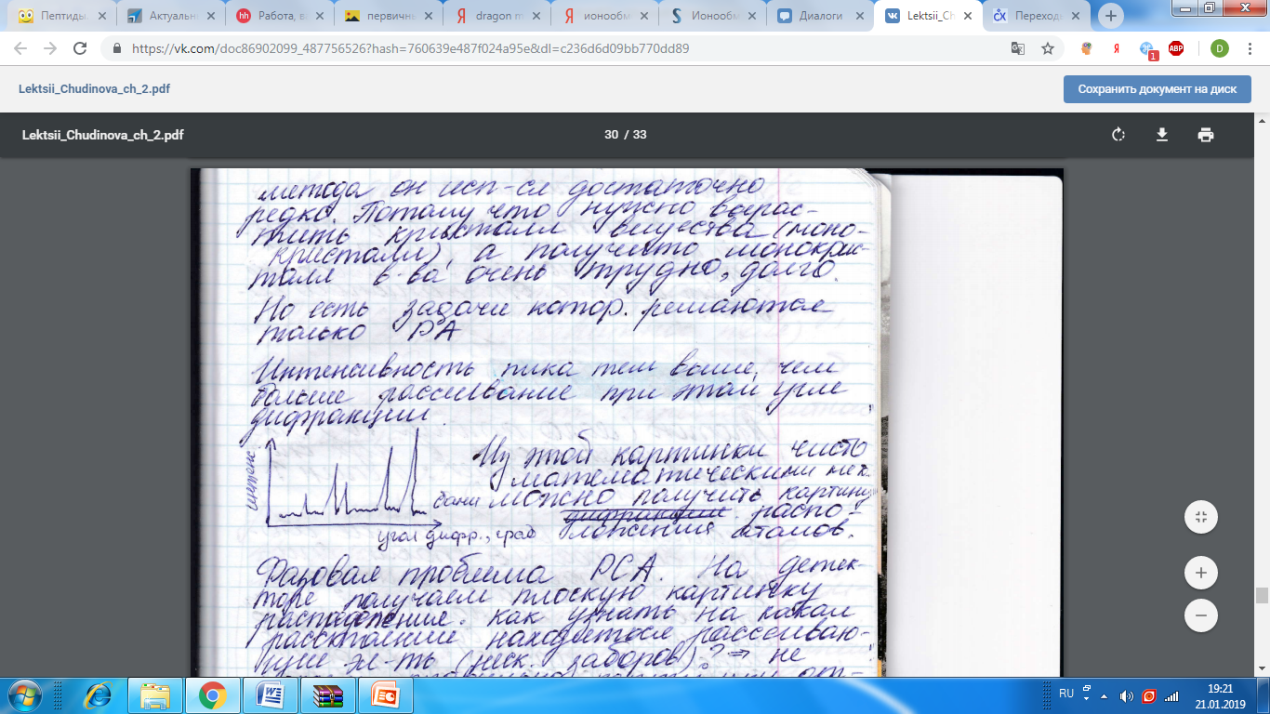
Первоначально поток излучения идет по центру, а регистрируем под углом.
Дифракция рентгеновских лучей – рассеяние рентгеновских лучей кристаллами (или молекулами жидкостей и газов), при котором из начального пучка лучей возникают вторичные отклоненные пучки той же длины волны, появившиеся в результате взаимодействия первичных рентгеновских лучей с электронами вещества (направление и интенсивность вторичных пучков зависят от строения рассеивающего объекта)
Кристалл – образец исследуемого вещества, в котором много (1012) идентичных молекул находятся в одинаковой ориентации и их центры образуют правильную трехмерную решетку (его размер не менее 1 мм, иначе невозможно работать)
σ
σ0



рентгеновское излечение


Симметрия, наблюдаемая на рентгенограммах, полностью соответствует симметрии исследуемых кристаллов
Принцип рентгеноструктурного анализа: по известной длине рентгеновского излучения определяется межплоскостное расстояние d, характеризующее структуру кристалла
Принцип рентгеноспектрального анализа: по известному межплоскостному расстоянию d определяются длины волн, входящих в состав рентгеновского излучения
Применение в бТ
Одним из важнейших экспериментальных методов, позволяющих с атомарной точностью узнать, что представляет собой трехмерная структура белка, т.е. определить пространственные координаты всех атомов исследуемого объекта, является рентгеноструктурный, или кристаллографический, анализ. Зная положение каждого атома, можно вычислить межатомные расстояния, валентные углы, углы вращения вокруг связей, распределение поверхностного заряда и другие детали молекулярной геометрии.
«+»:
-
В отличие от ЯМР РСА может доказать оптическую конфигурацию, так как рентген показывает расположение атомов в молекуле (как они взаимно друг к другу расположены)
-
Дешевый метод
-
Простой
«-»:
-
Необходимо выращивать монокристалл вещества, а это долго и трудно
-
Трудно из полученной плоской картинки узнать структуру вещества (расстояние между атомами), для этого в структуру добавляют несвойственные веществу атомы
Кристаллографические методы:
-
Классическая кристаллизация
-
Кристаллизация в условиях микрогравитации
-
Кристаллизация в микрофлюидах
-
Сокристаллизация белка с лигандом
Нативный белок Изоморфное производное




Тяжелые атомы

Применение: изучение структуры витаминов, антибиотиков, белков, полимеров, установление или уточнение химической формулы, типа связи, молекулярного веса при известной плотности или плотности при известном молекулярном весе, симметрии и конфигурации молекул и молекулярных ионов.
Для проведения рентгеновского эксперимента необходимо монохроматическое рентгеновское излучение (т.е. строго определенной длины волны). Для этой цели используются различные фильтры и монохроматоры.

Итак, на входе мы имеем неизвестный объект, на выходе – набор интенсивностей рассеянных в различных направлениях лучей, или дифракционную картину. Теперь необходимо связать полученную в эксперименте информацию с атомной структурой исследуемого объекта. Перечислим основные положения, на которых строится простейшая математическая модель рассеяния рентгеновских лучей:
1) пучок рентгеновских лучей является плоской монохроматической электромагнитной волной; 2) под воздействием этой электромагнитной волны каждый электрон приходит в движение, которое может быть описано уравнениями для свободных зарядов; 3) движущийся электрон является, в свою очередь, источником новой рассеянной сферической электромагнитной волны, распространяющейся во всех направлениях; 4) эти новые волны суммируются и определяют интенсивность излучения в интересующем нас направлении.
Метод рентгеноструктурного анализа основан на дифракции рентгеновских лучей на кристаллической решетке и поэтому применим только к веществам в кристаллическом состоянии. Это связано с тем, что для регистрации дифракционной картины рассеяния необходимо иметь достаточное количество рассеивающих электронов
РСА для изучения молекул основан на дифракции рентгеновских лучей кристаллами (или молекулами жидкостей/газов) при котором из начального пучка лучей возникают вторичные отклоненные пучки той же длины волны, появившееся в результате взаимодействия первичных рентгеновских лучей с электронами вещества, направление и интенсивность вторичных пучков зависит от строения рассеивающего объекта
Таким образом, центральная проблема метода рентгеноструктурного анализа, называемая фазовой проблемой, заключается в невозможности получения всех необходимых для расчета данных непосредственно из эксперимента.
Общего решения фазовой проблемы на сегодня не существует. Каждый случай требует специального подхода. Здесь важно понимать, что новая информация не берется ниоткуда. Для того чтобы получить значения фаз, мы должны либо сделать какие-то новые предположения о структуре и особенностях объекта, либо провести новые эксперименты. Ниже приведены основные подходы к решению «фазовой проблемы», применяемые в белковой кристаллографии.
Изоморфное замещение
Можно попытаться внедрить в молекулы кристалла некую метку – один или несколько тяжелых атомов (например, ионы тяжелых металлов), которые могут быть либо добавлены к нативной структуре, либо могут замещать часть ее атомов (рис. 5).

Рис. 5. Изоморфное замещение белков
Под изоморфным внедрением тяжелых атомов подразумевается, что они присоединяются к каждому экземпляру молекулы в одном и том же месте, и структура молекулы белка при этом не изменяется. Затем, проведя дополнительно рентгеновский эксперимент с таким модифицированным соединением и определив изменения интенсивностей рефлексов по сравнению с нативным белком, можно получить дополнительную информацию о значениях фаз. Трудность этого метода заключается в том, что не всегда удается получить хорошее изоморфное производное, а также в необходимости проведения дополнительного рентгеновского эксперимента.
Метод изоморфного замещения является основным методом решения фазовой проблемы при определении структуры биологических макромолекул. Сам этот метод возник достаточно давно, но именно при работе с белками он приобрел исключительно важную роль. Причин этому две:
1) долгое время он являлся единственным методом, позволяющим решать фазовую проблему для белков;
2) именно для белков удается «достаточно просто» получать изоморфные производные. Последнее связано с тем, что кристаллы белка довольно рыхлые – в них от 30 до 70% объема занято растворителем, т.е. в кристаллах есть «пустоты», куда могут поместиться дополнительные атомы.
Использование эффекта аномального рассеяния
Этот метод основан на варьировании длины волны падающего на кристалл рентгеновского излучения вблизи значений, при которых наблюдается эффект резонанса (и соответствующее аномальное рассеяние) для нескольких «специальных» атомов, содержащихся в структуре макромолекулы. Если аномально рассеивающих атомов в белке нет, иногда можно попытаться присоединить их химическим путем. Дифракционные картины получают для нескольких значений длины волны падающего луча и на основании анализа разностей интенсивностей соответствующих рефлексов оценивают значения фаз.
Успех метода аномального рассеяния, как и изоморфного замещения, во многом зависит от возможности экспериментального получения производных с требуемыми свойствами.
Упомянутые два способа отвечают попытке решить фазовую проблему за счет дополнительной информации, получаемой из дополнительных экспериментов. Следующий способ применяют в ситуации, когда нам известна структура близкого (гомологичного) белка.
Метод молекулярного замещения
В биологии распространена ситуация, когда существуют ряды объектов, похожих друг на друга, т.е. имеющих структурную гомологию. Такой гомологией могут обладать, например, белки одного типа, выделенные из разных организмов. В этом случае можно надеяться, что фазы структурных факторов, рассчитанные по известной атомной модели гомологичного белка, будут достаточно хорошим начальным приближением к значениям неизвестных фаз, отвечающих исследуемому объекту. Комбинируя их далее с измеренными в эксперименте модулями структурных факторов для исследуемого объекта, мы можем получить хорошее приближение к искомому распределению электронной плотности.
Однако для того чтобы надеяться на успех на этом пути, надо, как минимум, для начала «разместить» известный гомологичный объект на том же месте и в той же ориентации, что и исследуемый белок. Процедуру создания такого «компьютерного гибрида», в котором внутри элементарной ячейки кристалла одного белка размещается молекула другого, называют методом молекулярного замещения. Судить о том, насколько полученное размещение близко к действительности, можно, сравнивая рассчитанные по модели модули структурных факторов с величинами, полученными в эксперименте. Разумеется, такое замещение – всего лишь умозрительная процедура, и никакого химического замещения не происходит.
«Прямые» методы
В отличие от предыдущих подходов, эти методы опираются не на дополнительный эксперимент или информацию о структуре гомологичного объекта, а на почти философскую идею об атомности изучаемого объекта. Под «прямыми» методами в кристаллографии понимаются стратегии определения структур, использующие в качестве стартовой информации только набор интенсивностей рефлексов, полученный в рентгеновском эксперименте. Для определения фаз структурных факторов в них используют вероятностный подход. «Прямые» методы более объективны в том смысле, что они зависят только от применения математических соотношений.
На основе «прямых» методов определяют структуры большинства низкомолекулярных соединений. Эти методы не требуют ни дополнительных экспериментов, ни тонкой биохимической работы по получению изоморфных производных, ни наличия известных гомологичных структур, но к сожалению, пока не применимы к структурам белков из-за принципиальных ограничений на количество атомов исследуемой структуры.
Если
известны и модуль, и фаза структурных
факторов, то мы можем восстановить
распределение ![]() (r),
рассчитав обратное преобразование
Фурье. Это не сложная с современной
точки зрения вычислительная задача, и
этот шаг выделяется потому, что он
подводит итог важного этапа работ. Мы,
наконец, получаем возможность «взглянуть»
на интересующий нас объект. И по тому,
насколько «четким» получилось изображение,
– судить об успешности всех предыдущих
этапов работы. А в случае неудачи –
повторить все сначала.
(r),
рассчитав обратное преобразование
Фурье. Это не сложная с современной
точки зрения вычислительная задача, и
этот шаг выделяется потому, что он
подводит итог важного этапа работ. Мы,
наконец, получаем возможность «взглянуть»
на интересующий нас объект. И по тому,
насколько «четким» получилось изображение,
– судить об успешности всех предыдущих
этапов работы. А в случае неудачи –
повторить все сначала.
Следующий этап заключается в построении приближенной атомной модели по рассчитанным картам распределения электронной плотности. Эта работа требует максимального использования интеллекта человека и осуществляется квалифицированными специалистами.
С помощью специальных компьютерных программ, исследователь вручную вписывает атомы белковой структуры в полученную на предыдущем этапе карту электронной плотности (рис. 6).
http://bio.1september.ru/article.php?id=200900401