

43.Ректификация. Сущность ректификации. Устройство и принцип действия ректификац.Колонны. Типы тарелок, их преимущ-ва и недостатки.
Ректификация-физич.способ переработки нефти.
СУЩНОСТЬ РЕКТИФИКАЦИИ. РЕКТИФИКАЦИЯ - разделение жидких смесей на практически чистые компоненты, отличающиеся температурами кипения, за счет многократного испарения жидкости и конденсации паров.
Для ректификации используют РЕКТИФИКАЦИОННЫЕ КОЛОННЫ – вертикальные аппараты высотой до 20 м диаметром до 6 м, с теплоизоляцией снаружи. Как все массообменные аппараты бывают двух видов – тарельчатые и насадочные. Внутри тарельчатой колонны имеется 10 – 40 перегородок – тарелок. Снизу имеется кипятильник для перевода жидкости в паровую фазу – как правило кожухотрубчатый теплообменник, нагреваемый водяным паром, при перегонки нефти в качестве кипятильника используется трубчатая печь. В верхней части имеется конденсатор для конденсации паров – тоже кожухотрубчатый теплообменник охлаждаемый водой или воздушный холодильник. Часть сконденсированных паров отбирается в виде дистиллата, остальная часть направляется на орошение колонны и называется флегмой.(см.рисунок в тетради)
**Что происходит в ректификационной колонне:Сверху вниз идет поток жидкости – флегмы. Она перетекает по переливным устройствам с верхних тарелок на низлежащие. Снизу вверх идет поток пара обусловленный кипячением части жидкости в кипятильнике. На каждой тарелке происходят процессы массообмена: из пара в жидкость переходят более высококипящие компоненты а из жидкости в пар переходят легкокипящие компоненты. Таким образом жидкость, стекающая вниз обогащается тяжелыми компонентами, а пар, идущий вверх обогащается легкими.
Четкость разделения можно увеличить увеличивая количество тарелок и флегмовое число – отношение количества флегмы к количеству отбираемого дистиллата.
Процессы, проходящие в насадочных ректификационных колоннах аналогичны.
*Тарелки бывают различной конструкции.
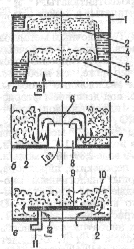
СИТЧАТЫЕ ТАРЕЛКИ (рис.первый) имеют перфорированное плато с диаметром отверстий 0,8-20 мм. Для них характерно динамическое взаимодействие газа с жидкостью, при котором «провал» отсутствует и реализуется ее переток по плато.
КОЛПАЧКОВЫЕ ТАРЕЛКИ (рис.второй) имеют колпачки разл. формы, снабженные прорезями в виде зубцов, проходя между которыми, газ (пар) диспергируется, что увеличивает поверхность его контакта с жидкостью. Эти тарелки также работают в беспровальном режиме и характеризуются более широким по сравнению с ситчатыми тарелками диапазоном нагрузок по фазам.
КЛАПАННЫЕ ТАРЕЛКИ (рис.третий) позволяют изменять свободное сечение установкой на их плато подвижных круглых или прямоугольных клапанов. Высота их подъема увеличивается с ростом скорости газа и регулируется спец. ограничителями либо весом клапана.
53.Производство мономеров для синтеза ск и пластмасс: стилора, бутадиена, изопрена.
СТИРОЛ: для синтеза стирола в кач-ве сырья прим-ют бензол. Первая стадия синтеза-получение этилбензола алкилированием бензола этиленом. Катализ-м при этом служит безводный AlCl3, активир-ный небольш.кол-вом HCl:
C6H5-H+CH2=CH2 = C6H5-CH2-CH3 (этилбензол);
C6H5-CH2-CH3+CH2=CH2 = С6Н4(С2Н5)2(диэтилбензолы)+Q
Реакция алкилирования обратимая. Она протекает далее с образованием изомерных диэтилбензолов, которые выделяют и добавляют в реакцион. смесь, чтобы сместить равновесие и повысить выход этилбензола. Для этого же применяют избыток бензола (2 моль бензола на 1 моль этилена). Вследствие большой активности кат-ра алкилир-е проводят при сравнительно низк. темп-ре (85—95 °С) и давлении, близком к атмосферн. Бензол и этилен, тщательно очищенные от примесей и высушенные, а также раствор катализатора в смеси диэтилбензолов вводят снизу в реактор—стальную эмалированную колонну высотой 10 м и диаметром 1,5 м; сверху из реактора отводится реакционн. смесь, которую промывают раствором щелочи и разделяют ректификацией на бензол, вновь направляемый в реактор, этилбензол (темп. кип. 136 °С) и смесь диэтилбензолов. Выход этилбензола до 95%, считая на этилен.
Во второй стадии синтеза стирола осуществляют дегидрирование этилбензола с образованием стирола: С6Н5 — СН2 — СН3 = С6Н5 — СН = СН2 + Н2 — Q
Здесь применяется железохромовый катализатор (Fe203+ Сг2Оз+К2О) или катализатор, состоящий из оксида цинка с добавлением СаО, K2SO4 и К2Сг04. В присутствии большого количества перегретого пара при 600°С превращение этилбензола достигает 45%. После конденсации паров и отделения воды смесь углеводородов разделяют ректификацией на бензол и толуол, образующиеся в результате крекинга, этилбензол (добавляемый к свежему этилбензолу) и стирол. Во избежание полимеризации стирола к смеси добавляют ингибитор — гидрохинон, а ректификацию производят в вакууме. Выход стирола до 90% (считая на этилбензол), темп. кип. 145°С. Производительность установки до 125 тыс. г в год.
БУТАДИЕН(дивинил): в 1928г С.В.Лебедев разраб-л промышлен.способ получения бутадиена-1,3 из этилового спирта. С. В. Лебедев нашел, что, применяя смешанный катализатор, можно одновременно осуществить как дегидратацию, так и дегидрирование с получением бутадиена:
2СН3-СН2ОН ------ СН2=СН-СН=СН2 + 2Н20 + Н2 — Q (смешанный катал-р: ускоряющих реакции дегидратации (А1203, Th02 и др.), легко отщепляется вода от этилового спирта с образованием этилена; дегидрирующими катализаторами (ZnO, Sn02 и др.)
Эта реакция, сопровождаемая многими побочными реакциями, осуществляется при 370—380 °С и давлении, близком к атмосферному, в контактной печи 3 (рис. 81). Печь имеет форму цилиндра высотой и диаметром до 6,5 м, выложена из огнеупорного кирпича с двойными стенками, внутри которой по окружности установлены 24 стальные реторты 4 прямоугольного сечения, заполненные катализатором. Кольцевое пространство между двойными стенками представляет собой топку 5, в которой сгорает газообразное топливо. Реторты нагреваются за счет лучеиспускания раскаленной внутрен. стенки. Топочные газы поступают во внутрен. пространство печи и далее в боров. Спирт-сырец смешивают с отходом производства-спиртом-регенератом, шихта испаряется в трубках спиртоиспарителя (на рис. 81 не указан). Пары спирта, проходя через перегреватели 1 и 2, омываемые топочными газами, нагреваются до 380⁰С, поступают в контактную печь 3 и здесь
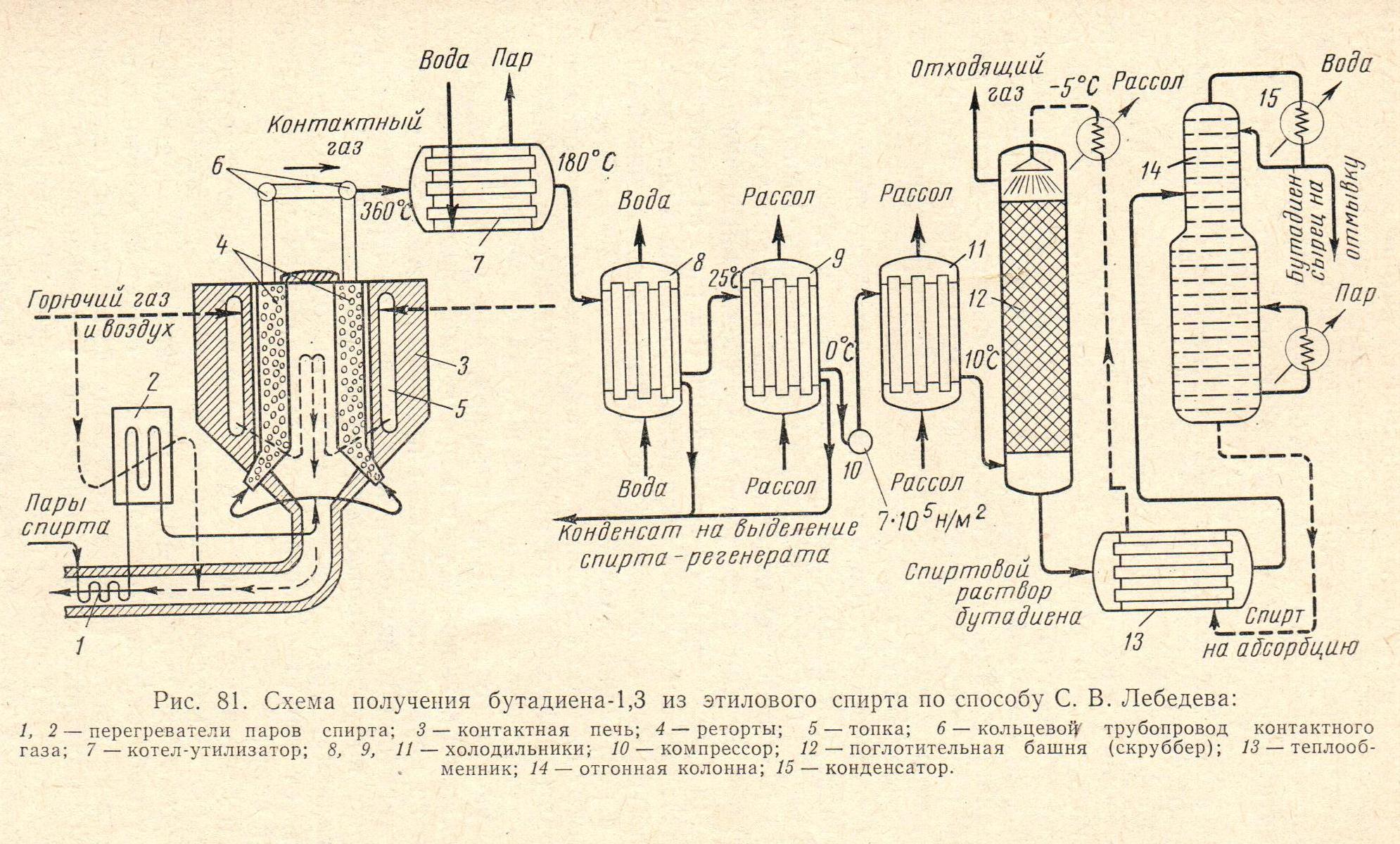
распред-ся по всем ретортам. Выходящий из реторт контактный газ собирается в кольцевом трубопроводе 6 и охлаждается до 180 °С в котле-утилизаторе 7. Газ представляет собой смесь, содержащую более тридцати веществ; помимо непрореагировавшего спирта и продуктов основной реакции — бутадиена, паров воды и водорода,— в ней содержатся в наибольшем количестве уксусный альдегид, диэтиловый эфир, этилен, пропилен и псевдобутилен (бутен-2).
Бутадиен — газ с темп-рой кипения — 4,5 °С, хорошо раств-ся в спирте и почти не раств-ся в воде и в разбавл-м водном спирте. Для его выделения и очистки прежде всего удаляют из контактного газа вещества с относительно высокой температурой кипения. Для этого газ охлаждают в холодильнике 8 водой, а затем в холодильнике 9 до 0°С холодильным рассолом. Конденсат — это водный спирт, который содержит эфир, уксусный альдегид и др. Ректификацией из него выделяют спирт-регенерат.
След. стадия очистки — извлечение спиртом бутадиена из газа. Для повышения раствор-ти и темп-ры кипения бутадиена газ предварительно сжимают до 7×105 н/м2 в компрессоре 10. После охлаждения нагревшегося при этом газа он поступает в три последовательно соединенные поглотительные башни (скруббера) 12 с насадкой (на рис. 81 изображен лишь один), где происходит абсорбция бутадиена холодн. спиртом, движ-ся противоточно движению газа. Отгонкой из р-ра в ректификац. колонне 14 получается бутадиен-сырец, а спирт из этой колонны после охлаждения в теплообмен-ке 13 вновь направляется на абсорбцию. Дальнейшая стадия очистки — удаление содер-ся в бутадиене-сырце этилов. спирта, уксусн. альдегида и эфира, для чего используется их хорошая растворимость в воде. В промывной колонне с насадкой из керамических колец примеси растворяются в воде при противочточном движении жидкостей. Ректиф-ей отмытого сырца получают бутадиен-ректификат с содер-ем 91—95% бутадиена; остальное — бутен-2. Выход бутадиена достигает 60% от теоретич. коли-ва, считая на прореаг-ший спирт.
***В настоящее время осуществлен новый способ получения бутадиена — катал-ким двухстадийным дегидрированием н-бутана, где сырьем служит бутановая фракция, выделяемая из попутного нефтяного газа, а также фракция н-бутана, получаемая в качестве отхода в производстве алкилата; сырье подвергается дополнительной ректификации для удаления примесей. В первой стадии дегидрирования образуется смесь изомерных бутиленов — бутена-1 и бутена-2:
СН3СН2СН2СНз(н-бутан) =С4Н8 + Н2 — Q (бутилены)
Для быстрого проведения процесса во избежание усиления побочных реакций крекинга применяется катализатор — оксид хрома на носителе-оксиде Al; активатором служит оксид К. При оптимальн. темп-ре 580°С и атмосферн. давлении равновесие достигается за 2 сек с превращ-ем 40% н-бутана в бутилены. Кат-р постепенно покрывается коксом и теряет свою активность. Применяется процесс с «кипящим» слоем пылевидного кат-ра, который сходен с процессом катал-го крекинга нефтепродуктов. В установку для дегидрирования также входят трубчатая печь для нагревания бутана, реактор и регенератор (оба с «кипящим» слоем катализатора). Выходящий из реактора контактный газ освобождается в циклоне от пыли кат-ра, затем постепенно охлаж-ся в котле-утилизаторе и в скруббере, орошаемом водой. Для того чтобы осуществить циркуляцию непрореаг-го бутана, необходимо его отделить от образ-ся бутиленов, водорода и продуктов побочн.реакций. Газ сжимают до 1,3-106 н/м2 и охлаж-т водой; выделившуюся при этом тяжелую фракцию (углеводороды С5 и выше) используют для извлечения из газа противоточной абсорбцией в колонне С4-фракции; затем ее выделяют из раствора ректификацией и конденсацией паров. Отделить бутан от бутиленов непосредственно ректификацией не удается вследствие близости темп-ур кипения. Но при введении в смесь ацетонитрила CH3CN (побочного продукта в производстве акрилонитрила) летучесть бутиленов уменьшается вследствие их лучшей растворимости в ацетонитриле по сравнению с летучестью бутана, который удаляется ректификацией. Этот способ разделения называют экстрактивной ректификацией. Раствор бутиленов из первой ректификац-й колонны поступает во вторую, отгонную колонну, в которой ректификацией пары бутиленов отделяются от менее летучего ацетонитрила. Выход бутиленов на прореаг-ший бутан составляет около 70%.
Бутилены во второй стадии дегидрирования превращаются в бутадиен на поверхности катализатора Са8Ni(Р04)6 + Сг20з:
С4Н8 = СH=СН-СН=СН2 + Н2 — Q (бутадиен-1,3)
В этой обратимой реакции при 620°С и атмосферном давлении превращению подвергается лишь 15% бутиленов; так как объем газа при протекании реакции возрастает, то уменьшение давления может повысить превращение до 40%. Понижение парциальных давлений углеводородов достигается введением в смесь больших кол-ств перегретого водяного пара (20 объемов на 1 объем бутиленов), который доставляет, кроме того, теплоту, необходимую для поддержания пост-ной темп-ры. Парогазовая смесь пропускается через неподвижный слой катализатора в виде шариков и затем из нее выделяется, как указано выше, фракция С4-УглВод-в, содержащая бутилены и бутадиен. Вследствие близости темп-р кипения разделить их ректиф-ей невозможно. Для их разделения используется способность холодного аммиачного раствора ацетата меди (I) извлекать из этой смеси бутадиен с образованием водораств-ого комплексного соединения. При нагревании из него выделяется бутадиен, а непоглощенные раствором бутилены направляются повторно на дегидрирование. Применяют также и экстрактивную ректиф-ю с ацетонитрилом. После дальнейшей очистки от примеси ацетилена и ректиф-и получают 99,5—99,8-процентный бутадиен с выходом до 55% на н-бутан. Себестоимость его ниже, а кач-во выше, чем полученного из спирта, ресурсы же н-бутана значительны. Поэтому сейчас уже более половины бутадиена производится этим способом. Производительность установки до 90 тыс. т в год.
ИЗОПРЕН: Аналогично и с теми же катализаторами проводят двухстадийное дегидрирование изопентана для получения изопрена. Изопентан выделяют из газов нефтепереработки, газового бензина и головной фракции, получаемой при подготовке бензина для его ароматизации; кроме того, выделенный из тех же продуктов нефтепереработки н-пентан подвергают каталит-кой изомеризации пропусканием паров при 400°С над кат-ром каталитич. риформинга, превращая его в изопентан.
В первой стадии дегидрирования образуется смесь трех изоамиленов: 2-метилбутена-1; 2-метилбутена-2 и 2-метилбутена-З с преобладанием второго из них:
С5H12 (изопентан) = С5Н10 (смесь изоамиленов) + Н2 — Q
При 540 °С степень превращения достигает 35% при селективности 70%; после этого выделяется Cs-фракция и из нее посредством экстрактивной ректификации с диметилформамидом НСОN(СН3)2 отгоняется непрореаг-ший изопентан, а из остатка выделяется смесь изоамиленов. Все они во второй стадии дегидрирования превращаются в изопрен (при 620 °С на 35%, с селективностью до 85%):
С5Н10 = СН2=С-СН = СН2 + Н2 - Q
СН3
Выделение изопрена из сложной смеси осуществляется экстрактивной ректиф-ей с диметилформамидом, после этого его дополнительно очищают ректиф-ей и избирательным гидрированием ацетилена и получают (с выходом до 55%) 99,5— 99,8-процентный изопрен (жидкость с темп. кип. 34 °С). Производительность установки достигает 120 тыс. г в год.