

Фармацевтическая химия - наука, которая изучает вопросы, связанные с лекарственными веществами: их получение и химическую природу, состав и строение, влияние отдельных особенностей строения их молекул на характер действия на организм, изучает физические и химические свойства лекарственных веществ и методы контроля их качества, определяет условия хранения лекарств.
Фармацевтическая химия занимает ведущее место в комплексе смежных фармацевтических наук (технология лекарств, токсикологическая химия, фармакогнозия, экономика и организация фармацевтического дела) и является необходимым фундаментом для их понимания и знания.
В то же время фармацевтическая химия, являясь специализированной наукой, не может не опираться на знания смежных химических (неорганическая, органическая, аналитическая, физическая и коллоидная химия), а также медико-биологических (фармакология, физиология, биологическая химия) дисциплин.
Знание биологических дисциплин необходимо для понимания сложных физиологических процессов, происходящих в организме, в основе которых лежат химические и физические реакции. Это позволяет более рационально применять лекарственные вещества, наблюдать за их действием в организме и на основании этого изменять в необходимом направлении структуру молекул создаваемых лекарственных веществ с целью получения желаемого фармакологического эффекта. По результатам фармакологического испытания лекарственных веществ дается заключение о возможности использования их в медицинской практике.
Действие лекарственного вещества определяется не только его химической структурой, но зависит также и от его физико-химических свойств. Поэтому фармацевтическая химия тесно связана с физической и коллоидной химией. Изучение структуры молекулы лекарственного вещества, разработка методов синтеза и анализа его невозможны без знания органической и аналитической химии. Вопросы совместимости лекарственных веществ в рецептурной прописи, способы изготовления, сроки годности, условия хранения и отпуска лекарств связывают фармацевтическую химию с технологией лекарств, экономикой и организацией фармации. Но решать вопросы совместимости, условия хранения лекарств может лишь специалист, владеющий знаниями фармацевтической химии.
На современном этапе фармацевтическая химия тесно связана с физикой и математикой. На общих законах этих наук базируются физико-химические методы анализа лекарств и расчеты в фармацевтическом анализе.
Правила выбора названия ЛС. Название ЛС – уникальное словесное обозначение ЛС в виде определенного сочетания букв или слов, соответствует установленным правилам его формулирования и выбора, а также названия предложенные его производителем.
Международное непатентованное наименование (МНН) – идентификация ЛС или его активной субстанции, путем присвоения ему укороченного наименования активной субстанции ЛС ВОЗ. Идентификатор фарм. субстанции или активного ингредиента. Название, используемое в официальной регламентации для лек. препаратов, эти названия не могут быть зарегистрированы как официальные.
Генерическое наименование – международные непатентованные наименования присваиваемые ВОЗ активному веществу предназначенные для использования в качестве общественной собственности без всяких ограничений, поскольку никто не может являться владельцем прав на его применение.
Торговое название ЛС – уникальное словесное ЛС в виде определенного сочетания букв или отдельных слов, под которым оно прошло гос. регистрацию, рекламируется и реализуется на фарм. рынке и может быть запатентовано.
Патентованное название ЛС – запатентованное торговое название ЛС охраняемое патентным законом государства
Анатомо-терапевтическо-химическая система классификации (АТХ) осуществляется работой ВОЗ. Данная система классифицируется по группам на 5 различных уровнях. 1 уровень: 14 основных анатомических групп. 2 уровень: основная терапевтическая группа. 3 уровень: терапевтически-фармакологическая подгруппа. 4 уровень: .5 уровень: подгруппа химических веществ. А (пищеварительный тракт, обмен веществ), В (органы кроветворения), С (сердечно-сосудистая система). Например: А01 – стоматологические препараты, А01АА – для профилактики кариеса, А01АА01 – фторид натрия.
по особенностям химического строения (например, производные фенотиазина, препараты лития, нитраты и нитриты); особенностям фармакологического действия (например, спазмолитики, бронхолитики); месту приложения эффекта (например, «петлевые» диуретики - мочегонные ЛС, действующие в почках на уровне петли Генле); особенностям механизма действия (например, неполяризующие мышечные релаксанты, необратимые ингибиторы моноаминоксидазы, избирательные ингибиторы нейронального захвата); особенностям терапевтического действия или нозологическим формам (например, ЛС для лечения нарушений сердечного ритма, язвенной болезни желудка и двенадцатиперстной кишки). Возможные комбинированные классификации ЛС приведены, например, в справочниках академика М. Д. Машковского «Лекарственные средства» или С. А. Крыжановского и М.Б.Вититновой «Современные лекарственные препараты».
Если в отношении классификаций не существует единого подхода, то названия (наименования, номенклатура) ЛС строго упорядочены и могут быть трех видов:1) полное химическое название ЛС. Химические названия лекарственных средств в широкой медицинской практике не используют. Они приведены только в специализированных справочных изданиях;2) непатентованное международное название, т.е. единое официально принятое во всех странах мира название, которое утверждено государственными органами здравоохранения различных стран и используется в национальных и международных фармакопеях;3) коммерческое (патентованное), или фирменное, название, которое является коммерческой собственностью фирмы-изготовителя (ее торговой маркой), т.е. один препарат может иметь неограниченное количество названий.
Источники получения лекарственных средств.
Для получения неорганических ЛС используется минеральное сырье (природные источники). Например, для приготовления ЛС натрия хлорида (Natrii chloridum) NaCl используются природные растворы — воды озер и морей; калия хлорида (Kalii chloridum) КС1 — минералы: сильвинит КС1 • NaCl, карналлит КС1 • MgCl2•6Н20
Синтетические органические ЛС получают из продуктов переработки каменного угля, нефти, дерева, горючих сланцев. Выделенные при этом индивидуальные органические соединения являются реагентами в органическом синтезе лекарственных веществ. Так осуществлен полный химический синтез антибиотика левомицетина и алкалоида кофеина.
Источником получения органических лекарственных веществ является растительное лекарственное сырье. Из него получают алкалоиды, терпены, гликозиды, витамины, эфирные и жирные масла, белки, углеводы. Растительное сырье используют также для получения галеновых препаратов.
Гормональные препараты готовят из сырья животного происхождения (органы и ткани животных). Для получения антибиотиков используют различные микроорганизмы. Известны полусинтетические антибиотики, которые являются синтетическими производными антибиотиков, выделенных из микроорганизмов (например, пенициллины и цефалоспорины). Полусинтетический способ применяется для получения и других групп ЛС: алкалоидов, витаминов, гормонов, анаболических стероидных препаратов.
Способы создания новых ЛС: 1. новые случайные открытия, 2. скрининг, 3. наблюдение за побочными эффектами, 4. комбинаторная химия, 5. QSAR (метод Ганча, Фри Вилсена), 6. генная инженерия, 7. компьютерное и молекулярное моделирование, 8. метод направленного синтеза известных биообъектов.
Современные требования к ЛС.
Безопасность ЛС – положительная характеристика ЛС основанная на сравнительном анализе его эффективности и оценки риска причинения вреда жизни и здоровью человека. В качестве главного критерия определяющего допуск ЛС на фарм. рынок выступает показатель РИСК / ПОЛЬЗА
Эффективность ЛС – характеристика степени «+» влияния ЛС на предупреждение, течение или продолжительность заболевания, предотвращения беременности, восстановление нормальной жизнедеятельности организма человека и компенсацию его функциональных возможностей нарушенных в результате заболевания.
Качество ЛС – отечественного производства: определяется соответствием ЛС требования ФС производителя, ФС ГФРБ, а ЛС зарубежного производства: требованиям нормативного документа его производителя содержащим показатели и методы контроля за качеством ЛС
Обеспечение качества ЛС достигается за счет: 1. обучения и аттестации специалистов, работающих в области обращения ЛС, 2. разработка и внедрение надлежащих практик обращения с ЛС, 3. исключение из реестра ЛС сомнительной терапевтической эффективности.
Качество ЛС – совокупность характеристик относящихся к его способности удовлетворять установленные и предполагаемые потребности и состоящие из основных показателей: 1) безопасность (токсичность, побочные реакции), 2) эффективность, 3) фарм. качество (соотв. требованиям нормативного документа), 4) доступность.
Т. к. ЛС особая продукция, которая может нанести вред человеку (животному), то показатели качества не могут устанавливаться предпринимателем-производителем, они должны устанавливаться государством и государством контролироваться.
Надлежащие практики – совокупность правил по организации и по мере: доклинические исследования (GLP), клинические испытания (GCP), промышленное производство (GMP), оптовой реализации (GDP), аптечного изготовления и реализации (GPP), обеспечивает и гарантирует их качество и доступность.
Надлежащая лабораторная практика – совокупность правил по планированию, выполнению, контролю, оценке и документированию результатов доклинических исследований, осуществляемых при разработке новых лекарственных средств;
Надлежащая клиническая практика – совокупность правил по планированию, выполнению, контролю, оценке и документированию результатов клинических испытаний лекарственных средств;
Надлежащая производственная практика (GMP) – совокупность правил по организации промышленного производства и контроля за качеством ЛС
Надлежащая аптечная практика (GPP) – совокупность правил по контролю за качеством, сроку годности, упаковки и маркировки, условий хранения, а также реализации ЛС
Надлежащая практика оптовой реализации – совокупность правил по организации оптовой реализации лекарственных средств, обеспечивающих и гарантирующих их качество и доступность;
2006г ТКП (Технический Кодекс установл практики) 9 разделов 7 приложений
Организация контроля качества лекарственных средств в Республике Беларусь Данный контроль осуществляет Министерство здравоохранения через свои структурные подразделения. Практическую работу по контролю качества лс в РБ осуществляют Республиканская контрольно-аналитическая лаборатория, семь контрольно-аналитических лабораторий областных УП «Фармация», лаборатория научно-производственного государственного предприятия МБИ-ЛОТИОС, научно-исследовательский центр предприятия диагностических и лекарственных препаратов «Диалек» и Белорусский НИИ эпидемиологии и микробиологии. Качество лекарств, изготовляемых в аптеках, проверяется провизорами-аналитиками аптек, а их рабочим местом является аналитический кабинет либо аналитический стол. Новым звеном в этой схеме для РБ является «Республиканский центр экспертиз и испытаний в здравоохранении», который начал свою деятельность в 1998 году. Он создан в целях обеспечения безопасности лекарственных средств, изделий медицинского назначения, медицинской техники, товаров народного потребления, химических и биологических веществ, материалов и изделий из них, продуктов питания и продовольственного сырья, а также соблюдения правил. Стратегические вопросы номенклатуры ЛС и контроля их качества рассматриваются на Фармакологическом и фармакопейном комитетах МЗ. Фармакологический комитет занимается рассмотрением и утверждением материалов о целесообразности внедрения в медицинскую практику новых и исключении устаревших лекарственных средств. Через данный комитет проходят все материалы клинических испытаний предлагаемого нового лекарственного средства. Комитет утверждает перечень импортируемых лекарственных средств.Фармакопейный комитет руководит работой по составлению и утверждению нормативной документации на ЛС. Комитет занимается вопросами стандартизации методов анализа лекарственных средств, утверждает сроки годности, условия хранения, обеспечивает региональные лаборатории нормативной документацией для контроля качества импортных лекарственных средств и т.д..Важная роль в обеспечении необходимого качества лекарственных средств отечественного производства принадлежит отделам технического контроля (ОТК) фармацевтических предприятий. Главными задачами ОТК являются: предотвращение выпуска (поставки) предприятием продукции, не соответствующей требованиям ЦД, повышение ответственности всех звеньев производства за качество выпускаемой продукции.
Нормативная документация регламентирующая качество ЛС
ГФ РБ в 3 томах, носит законодательный характер.
Том 1 (2006) «Общие методы контроля качества ЛС» общие ФС и методы фарм анализа
Том 2 (2008) «Контроль качества вспомогательных веществ и ЛРС» частные ФС на субстанции…
Том 3 (2009) «Контроль качества вспомогательных веществ, ЛРС и экстемпоральных ЛС» ФС на изготовление твёрдых, жидких и других лек. форм
Фармацевтический анализ – наука о хим характеристике (структуре и свойствах) и измерении фармакологической (биологической) активных веществ на всех этапах производства от контроля сырья до готового лек вещества (ЛС), изучение его стабильности, установление сроков годности и стандартизации
Фарм анализ применяется на всех этапах жизненного цикла ЛС (от разработки до уничтожения)
Виды фарм анализа: 1) фармакопейный анализ, 2) постадийный контроль производства ЛС (лек веществ), 3) экспресс-анализ в условиях аптеки, 4) биофармацевтический анализ.
Особенности фарм анализа: 1) анализу подвергают вещества различной хим природы, 2) широкий диапазон концентраций анализируемых веществ, 3) как правило, анализируется смесь веществ
Фармакопе́я — сборник официальных документов (свод стандартов и положений), устанавливающих нормы качества лекарственного сырья — медицинских субстанций, вспомогательных веществ, диагностических и лекарственных средств и изготовленных из них препаратов. Государственная фармакопея является документом общегосударственной законодательной силы, его требования обязательны для всех организаций данного государства, занимающихся изготовлением, хранением и применением лекарственных средств, в том числе растительного происхождения.Первый том ГФ РБ содержит обязательные стандарты и положения, регламентирующие качество лекарственных средств: общие статьи на методы анализа (физические, физико-химические, биологические, определение подлинности, определение примесей, количественное определение, методы анализа лекарственного растительного сырья, фармацевтико-технологические испытания), контейнеры, реактивы, общие тексты (по стерилизации, информация о вакцинах, статистические методы обработки результатов анализа), общие статьи на лекарственные формы. Кроме того, в издание включен раздел определения сравнительной биодоступности и биоэквивалентности генерических лекарственных средств. Второй том содержит реактивы, общие статьи на лекарственные формы, частные статьи на субстанции для фармацевтического использования и лекарственное растительное сырье. Кроме того, в издании включен перечень опечаток, допущенных в 1-м томе. Третий том содержит обязательные стандарты и положения, регламентирующие качество лекарственных средств, общие статьи на методы анализа (физические и физико-химические методы, испытания на предельное содержание примесей, биологические испытания, фармацевтико-технологические испытания), реактивы, общие статьи на лекарственные формы, частные статьи на субстанции для фармацевтического использования и лекарственное растительное сырье.
Национальные фармакопеи. Многие страны имеют собственные фармакопеи. Всемирная организация здравоохранения издаёт Международную Фармакопею, не имеющую, однако, законодательного характера, в отличие от национальных фармакопей. В странах, не имеющих собственных фармакопей, используют международную, европейскую или другую. Многие химико-фармацевтические предприятия выпускают субстанции по требованию заказчика — в соответствии с заданным стандартом, с той или иной фармакопеей.
Международная Фармакопея — первый том первого издания опубликован в 1951 г., второй том — в 1955 г. В 2006 г. увидело свет четвертое издание в двух томах. В 2008 г. оно же переиздано вместе с первым дополнением. Этой Фармакопеей пользуются страны, не имеющие национальную фармакопею.
Структура системы обеспечения безопасности, эффективности и контроля качества: 1) МЗ РБ, 2) Управление по лицензированию мед и фарм деятельности при МЗ РБ, 3) Комиссия по ЛС МЗ РБ, 4) РУП «Центр экспертиз и испытаний в здравоохранении», при котором организованы 3 лаборатории: 1. республиканская КАЛ, 2. лаборатория фармакопейного анализа, 3. клинико-фармакологическая лаборатория. Также существует 6 областных КАЛ, Лаборатория Минского аптечного склада.
Основные направления деятельности: 1) разработка нормативных документов регулирующих обращение ЛС на территории РБ, 2) контроль за соблюдением правил проведения испытаний ЛС на животных и людях, 3) аккредитация ЛПУ на право проведение клинических испытаний ЛС и аттестация специалистов по их проведению, 4) проведение гос регистрации ЛС (система учета и допуска к промышленному производству, реализации и мед применению ЛС произведенных в РБ, а также реализации и мед применению ЛС поступающих из-зи рубежа, которые призваны соответствовать тоебованиям по безопасности, эффективности и качеству), 5) проведение гос сертификации ЛС, 6) проведение стандартизации ЛС, 7) осуществление фармаконадзора (учет и анализ побочных реакций ЛС), 8) определение порядка регистрации БАД к пище, 9) лицензирование фарм деятельности, 10) инспектирование фарм деятельности, 11) контроль качества ЛС, 12) информационная работа, 13) международное сотрудничество и гармонизация требований к ЛС в других странах
В случае синтетического способа получения органических лекарственных веществ различают полный синтез и частичный синтез или полу синтез,
Полный синтез состоит из нескольких стадий ( их может быть и более 10), структура молекулы создается последовательно с использованием соответствующих исходных веществ, поставляемых химико-фармацевтическим предприятиям заводами химической промышленности.
Полусинтез включает 1-2 стадии, в процессе которых в структуру молекулы исходного вещества вносятся небольшие изменения (вводится, либо удаляется функциональная группа или радикал). Примерами могут служить получение кодеина из морфина, полу синтетических пенициллинов и цефалоспоринов.
В начале 40-ых годов появился новый способ получения лекарственных веществ - микробиологический синтез, который был предложен для получения пенициллина, в дальнейшем и других антибиотиков, а затем и некоторых витаминов, например, витамина Bi2. Микробиологический синтез в качестве отдельных стадий используется в схемах синтеза некоторых стероидных гормонов и получает все большее распространение, ибо позволяет осуществлять с большими выходами такие превращения, которые химическими методами произвести невозможно.
Последним достижением является промышленное освоение нового метода получения ранее трудно доступных лекарственных веществ на основе достижений генной инженерии. Этим методом уже получают инсулин, интерферон и др.
Титриметрическими называют методы анализа, основанные на титровании. Титрование — это процесс определения вещества, при котором к нему постепенно прибавляют небольшие порции реагирующего с ним другого вещества до того момента, пока всё определяемое вещество не вступит в реакцию. Реагент, используемый для титрования, называется титрантом. Обычно в титриметрических методах анализа титрант добавляют к анализируемому веществу в виде раствора с точно известной концентрацией растворённого вещества. Количество титранта, вступившего в реакцию, определяется по объёму раствора, затраченному для титрования. Момент титрования, при котором количество прибавленного титранта становится химически эквивалентным количеству определяемого вещества, называется точкой эквивалентности.В зависимости от типа химической реакции, протекающей между определяемым веществом и титрантом, выделяют: 1) кислотно-основное титрование - титриметрические методы анализа, основанные на протолитических реакциях; 2) комплексометрическое титрование - титриметрические методы анализа, основанные на реакциях образования растворимых комплексных соединений; 3) осадительное титрование - титриметрические методы анализа, основанные на реакциях образования малорастворимых соединений; 4) окислительно-восстановительное титрование - титриметрические методы анализа, основанные на окислительно-восстановительных реакциях.
Кислотно-основное титрование (протолитометрия) - это раздел титриметрии с использованием кислотно-основной реакции (реакции нейтрализации). Название этот раздел получил от протолитической теории кислот и оснований. Протолиметрию подразделяют на ацидиметрию (от лат. аcidum - кислота) и алкалиметрию (от лат. alcalum - щелочь). В ацидиметрии в качестве рабочего раствора используют титрованные растворы сильных кислот, а в алкалиметрии - щелочей. Прямым титрованием в протолиметрии определяют концентрацию кислоты или основания, или содержание элементов, образующих растворимые кислоты и основания (например фосфора в виде фосфорной кислоты и т.п.). Обратным или косвенным титрованием находят содержание некоторых солей. Например, для определения содержания NH4+ в NH4Cl обратным титрованием можно добавить к анализируемому раствору точно отмеренный избыток стандартного раствора NaOH, нагреть смесь до полного удаления NH3, а затем остаток раствора NaOH оттитровать кислотой в присутствии метилового оранжевого. Косвенный вариант титрования NH4+ можно осуществить формальдегидным методом, заместив ионы NH4+ эквивалентным количеством Н+ - ионов реакцией раствора NH4Cl с избытком формальдегида по реакции NH4Cl + 6CH2O (CH2) 6N4 + HCl + 6H2O Содержание NH4+ находят по результатам алкалиметрического титрования заместителя, т.е. HCl. Протолиметрическое титрование в основном проводят в водной среде, но существует и неводный вариант. В последнем случае, подобрав соответствующий растворитель, можно направленно изменять силу растворенных в нем кислот и оснований, превращать соли в кислоты и основания и т.д. Например, HCN в водном растворе - слабая кислота, а в среде сжиженного аммиака - сильная, мочевина в растворе безводной СН3СООН - сильное основание, а в сжиженном аммиаке - кислота и т.п. Поэтому применение неводного титрования делает возможным титрование очень слабых кислот и оснований, различных смесей солей с близкими свойствами, смесей солей с кислотами и основаниями, определение нерастворимых в воде и разлагаемых ею соединений. В зависимости от относительной силы кислот и оснований различают четыре основных случая протолиметрического титрования, каждый из которых моделируется соответствующей ТКТ: I - сильную кислоту титруют сильным основанием; II - сильное основание титруют сильной кислотой; III - слабую кислоту титруют сильным основанием; IV - слабое основание титруют сильной кислотой.
Определение азота в органических соединениях методом Кьельдаля проводят следующим образом. Точную навеску анализируемого образца помещают в колбу Кьедьдаля и подвергают минерализации с помощью концентрированной серной кислоты, к которой добавлены K2S04 и CuS04, а в некоторых случаях ещё и селен или HgO. В процессе окисления органической части молекулы азот восстанавливается до иона аммония. После окончания минерализации к раствору добавляют NaOH. При этом образуется NH3, который отгоняют и поглощают раствором Н3В03 или стандартным раствором сильной кислоты (H2S04 или НО). В первом случае при взаимодействии борной кислоты с аммиаком образуется эквивалентное NH3 количество иона В02, который затем титруют стандартным .раствором НС1 (титрование заместителя). Во втором случае определяют избыток сильной кислоты, не вступивший в реакцию с NH3, титруя раствор стандартным раствором NaOH (обратное титрование).
Обычный метод Кьельдаля используют для органических соединений, содержащих аминный азот (амины, аминокислоты и т.д.). Для определения азота в нитратах, нитритах, нитросоеди-нениях и т.п. необходимо ещё предварительное восстановление данных азотсодержащих групп до иона аммония или аминогруппы.
Методику, похожую на описанную выше, можно использовать также и для веществ, которые легко гидролизуются с образованием аммиака или аминов. Такие вещества не подвергают минерализации, а сразу проводят их щелочной гидролиз.
Ион аммония является достаточно слабой кислотой (рКа = 9,24), поэтому его прямое титриметрическое определение при концентрации и водном растворе, например, 0,1 моль/л, невозможно.
ОПРЕДЕЛЕНИЕ NH4: 1) формальдегидный способ (титрование заместителя) 4NH4+ + 6СН20 → (CH2)6N4 + 4Н+ + 6Н20 титруют стандартным раствором щёлочи в присутствии фенолфталеина
2) обратное титрование NH4+ + 0H- → NH3↑ + H20 избыток щелочи, не вступивший в реакцию, титруют стандартным раствором сильной кислоты
Окислительно-восстановительное титрование (редоксиметрия или оксидиметрия) основано на использовании реакций окисления-восстановления. Окислители титруют восстановителями, а восстановители – окислителями.
Требования к реакциям в Red-Ox-метрии. К реакциям, применяемым в окислительно-восстановительном титровании, предъявляются следующие требования: а) Реакции должны протекать до конца, являться необратимыми, не сопровождаться побочными процессами. б) В ходе реакции должны образовываться продукты определенного известного состава. в) Реакции должны протекать быстро. г) Должна быть возможность фиксировать точку эквивалентности.
К недостатком окислительно-восстановительных реакций в большинстве случаев относится их невысокая скорость, что затрудняет процесс титрования. Для ускорения реакций применяют нагревание. Если нагревание использовать нельзя (вещество разлагается или улетучивается), то увеличивают концентрацию вещества или используют катализаторы.
Классификация методов Red-Ox-метрии и способы титрования.Окислительно-восстановительное титрование или редоксиметрия (от латинского oxydatio - окисление и reductio - восстановление) основано на реакциях окисления-восстановления. Если титрант – окислитель, то титрование называют окислительным (оксидиметрия). Если титрант – восстановитель, то титрование восстановительное (редуциометрия)
По типу применяемого титранта методы окислительно-восстановительного титрования делятся на следующие виды:
• Пермангатометрическое – титрант раствор KMnO4;
• Иодометрическое титрование – титранты растворы I2 и Na2S2O3;
• Броматометрическое – титрант раствор KBrO3;
• Бромометрическое – титрант раствор Br2 (KBrO3+KBr);
• Хроматометрическое – титрант раствор K2Cr2O7 и т.д.
Редокс-титрование может быть выполнено различными способами: прямое титрование, обратное титрование и заместительное титрование.
Окислительно-восстановительные индикаторы. Для определения точки эквивалентности в Red-Ox-метрии используют различные индикаторы:
1) Окислительно-восстановительные индикаторы (редокс-индикаторы), изменяющие цвет при изменении окислительно-восстановительного потенциала системы.
2) Специфические индикаторы, изменяющие свой цвет при появлении избытка титранта или исчезновении определяемого вещества. Специфические индикаторы применяют в некоторых случаях. Так крахмал – индикатор на присутствие свободного йода, вернее трииодид-ионов I3-. В присутствии I3- крахмал при комнатной температуре синеет. Появление синей окраски крахмала связано с адсорбцией I3- на амилазе, входящей в состав крахмала.
Иногда в качестве индикатора используют тиоцианат аммония NH4SCN при титровании солей железа(III), катионы Fe3+ с ионами SCN- образуют соединение красного цвета. В точке эквивалентности все ионы Fe3+ восстанавливаются до Fe2+ и титруемый раствор из красного становится бесцветным.
При титровании раствором перманганата калия сам титрант играет роль индикатора. При малейшем избытке KMnO4 раствор окрашивается в розовый цвет.
Редокс-индикаторы делятся на: обратимые и необратимые.
Обратимые индикаторы – обратимо изменяют свой цвет при изменении потенциала системы. Необратимые индикаторы – подвергаются необратимому окислению или восстановлению, в результате чего цвет индикатора изменяется необратимо.
Редокс-индикаторы существуют в двух формах окисленной и восстановленной, причем цвет одной формы отличается от цвета другой.
Переход индикатора из одной формы в другую и изменение его окраски происходит при определенном потенциале системы (потенциале перехода).
Комплексонометрия (трилонометрия) — титриметрический метод, основанный на реакциях образования комплексных соединений ионов металлов с этилендиаминтетрауксусной кислотой и другими аминополикарбоновыми кислотами (комплексонами). Большинство ионов металлов взаимодействуют с комплексонами практически мгновенно с образованием растворимых в воде малодиссоциированных соединений постоянного состава. Метод позволяет определять практически все катионы и многие анионы. Комплексонометрия является составной частью комплексиметрии (хелатометрии).
Комплексонообразующими агентами, используемыми в качестве титрантов, являются аминополикарбоновые кислоты, имеющие характерную группу
Такие соединения способны образовывать хелатные комплексы со многими катионами, в которых катион связан в кольцевой структуре. Кольцо образовано солевыми связями катиона с карбоксильными группами, а также координационной связью за счет свободной пары электронов атома азота. Если кольцо шятичленное, то образованный хелат должен иметь высокую стабильность, так что наиболее удобные хелатные титранты это те, которые способны образовывать такие кольца. Это справедливо для этилендиаминтетрауксусной кислоты (ЭДТА), обычно 'применяемой в виде динатриевой соли, известной как едетат динатрия. С большинством металлов, имеющих более одного положительного заряда, ЭДТА образует высокорастворимые в воде комплексы при соотношении 1 : 1. При этом образуется структура, содержащая не менее 3 шятичленных хелатных колец, что обеспечивает высокую устойчивость комплекса. В некоторых случаях, помимо связей, которые образуются за счет свободной пары электронов азота, могут образовываться координационные связи за счет карбонильных кислородов других карбоксильных групп. Так, комплексы, образованные кальцием и трехвалентным алюминием, могут быть представлены формулами:
Стабильность таких комплексов в значительной степени зависит от рН раствора. Большинство двухвалентных металлов образует комплексы, устойчивые в щелочной среде, но хелаты щелочноземельных металлов 'разрушаются при рН примерно ниже 8; в то же время многие комплексы двухвалентных металлов (например, цинка и свинца) также устойчивы в достаточно кислом растворе. Комплексы трехвалентных металлов благодаря дополнительной стабильности, которая обеспечивается увеличенным числом хелатных колец, часто устойчивы даже в сильно кислых растворах. В щелочных растворах, однако, некоторые из этих металлов в присутствии ЭДТА осаждаются в виде гидроокисей, но не вследствие нестабильности комплекса, а вследствие более мощного влияния низкой растворимости гидроокиси металла.
Хелат алюминия образуется медленно, так что этот металл обычно определяют методом обратного титрования.
Для определения эквивалентной точки при титровании ионов металлов с ЭДТА необходимо использовать подходящий индикатор, который !будет реагировать на присутствие свободных ионов металлов в растворе. Таким индикатором, первоначально использованным Шварценбахом для титрования ионов кальция, был мурексид (пурпурат аммония), который в настоящее время очень редко применяется. Наиболее часто из индикаторов используется протравной черный 11 (известный также под другими торговыми названиями). Этот индикатор имеет синюю окраску в аммиачном растворе, но дает красные комплексы со многими ионами металлов в этом растворе. Образуемые при этом комплексы металлов, как правило, менее прочные, чем соответствующие комплексы с ЭДТА, так что титрование с эдетатами легко удаляет металл из его комплекса с индикатором и изменение цвета на чисто синий свидетельствует о полном титровании металла, имеющегося в растворе. Протравной черный 11 часто используют в смеси с метиловым оранжевым; на 'фоне последнего легче обнаруживается конечная точка титрования.
Другой широко применяемый индикатор — ксиленоловый оранжевый; это обычный кислотно-основный индикатор, в который введены группы иминодиуксусной кислоты, благодаря чему это вещество действует в качестве металл-комплексного индикатора. Этот индикатор дает четкий переход окраски от розово-фиолетовой к желтой в конце титрования алюминия, висмута, свинца, ртути и цинка и может применяться при рН 2—6 в зависимости от титруемого металла.
Осадительное титрование - группа титриметрических методов анализа, основанных на реакциях образования малорастворимых соединений, выделяющихся из раствора в виде осадка.Лишь немногие из реакций образования осадков пригодны для титриметрии. Для того чтобы реакция могла быть использована в осадительном титровании: 1) она должна протекать количественно (величина Ks осадка для бинарных электролитов не должна быть больше 10-8-10-10) и быстро при обычных условиях, не сопровождаться образованием пересыщенных растворов; 2) образующийся осадок должен иметь постоянный состав и не должен загрязняться в процессе осаждения; 3) должен существовать способ обнаружения конечной точки титрования.Среди всех методов осадительного титрования практическое значение имеют аргентометрическое и меркурометрическое титрование. Однако и данные методы, несмотря на большой круг возможных определяемых объектов среди лекарственных веществ, применяются нечасто.
Аргентометрическим титрованием называется титриметрический метод анализа, основанный на образовании малорастворимых соединений серебра.Основным титрантом в аргентометрии является AgNO3. Аргентометрия используется для определения галогенидов, тиоцианатов, цианидов, фосфатов и других ионов. Аргентометрию применяют для количественного определения галогенидов натрия и калия и некоторых органических галоген-производных (после их гидролиза в соответствующих условиях). Выбор метода обнаружения конечной точки аргентометрического титрования зависит от природы определяемого вещества, рН раствора и присутствия в нём других соединений. Среди различных вариантов аргентометрических определений чаще всего применяют метод Фольгарда. Метод Мора используется, в основном, для определения хлоридов и бромидов натрия и калия в нейтральных или слабощелочных растворах. Для определения галогенидов в присутствии катионов азотсодержащих органических соединений этот метод не подходит из-за того, что водные растворы большинства таких веществ имеют кислую среду. При попытке нейтрализовать раствор, органическое основание может выпасть в осадок. Метод Фаянса обычно используют для определения иодидов.Аргентометрия может быть использована также для определения органических веществ, образующих малорастворимые серебряные соли, например, барбитуратов, сульфаниламидов, теофиллина и др.Меркурометрическим титрованием называется титриметрический метод анализа, основанный на образовании малорастворимых соединений ртути (I).Меркурометрическое титрование используется, главным образом, для определения хлорид- и бромид-ионов.В качестве титранта в меркурометрии используют Нg2(NОз)2. Стандартный раствор этого вещества является вторичным. Для приготовления данного раствора навеску Hg2(N03)2-2H20 (х.ч.) растворяют при нагревании в -0,2 М HNCb. Для восстановления содержащейся в нём примеси Hg2+ до Hg2+ к раствору прибавляют металлическую ртуть (2-3 капли на литр растврра). Затем раствор хорошо взбалтывают и оставляют на сутки. После этого раствор фильтруют и к фильтрату вновь добавляют несколько капель металлической ртути. Для стандартизации приготовленного раствора применяют NaCl. Хранят стандартный раствор Hg2(N03)2 в склянках тёмного стекла в защищенном от света месте. ИНДИКАТОРЫ, ИСПОЛЬЗУЕМЫЕ В МЕРКУРОМЕТРИИ: 1) Дифенилкарбазон (адсорбционный индикатор, вносят как можно ближе к концу титрования) 0,2-5 М HN03. 2) Тиоцианатные комплексы Fe(III) (Для того чтобы учесть количество титранта, израсходованное для взаимодействия с индикатором, проводят контрольный опыт)Широкого распространения в практике фармацевтического анализа меркурометрическое титрование не имеет (в отличие от меркуриметрического). Его положительной характеристикой является возможность определения галогенид-ионов в сильнокислых растворах.
В абсорбционных методах излучение постороннего источника пропускают через пробу, при этом часть квантов избирательно поглощается атомами или молекулами. Важнейшие методы этой группы - атомно-абсорбционный анализ (ААС) и молекулярно-абсорбционная спектроскопия растворов. Последний метод обычно называют спектрофотометрией или фотометрическим анализом. Абсорбционные методы используют и для обнаружения, и для количественного определения веществ.
АБСОРБЦИОННАЯ СПЕКТРОСКОПИЯ, изучает спектры поглощения электромагн. излучения атомами и молекулами в-ва в разл. агрегатных состояниях. Интенсивность светового потока при его прохождении через исследуемую среду уменьшается вследствие превращения энергии излучения в разл. формы внутр. энергии в-ва и (или) в энергию вторичного излучения. Поглощат. способность в-ва зависит гл. обр. от электронного строения атомов и молекул, а также от длины волны и поляризации падающего света, толщины слоя, концентрации в-ва, т-ры, наличия электрич. и магн. полей. Для измерения поглощат. способности используют спектрофотометры-оптич. приборы, состоящие из источника света, камеры для образцов, монохроматора (призма или дифракционная решетка) и детектора. Сигнал от детектора регистрируется в виде непрерывной кривой (спектра поглощения) или в виде таблиц, если спектрофотометр имеет встроенную ЭВМ. Применение абсорбционной спектроскопии основано на след. законах.
1. Закон Бугера-Ламберта: если среда однородна и слой в-ва перпендикулярен падающему параллельному световому потоку, то I = I0 exp (— kd), где I0 и I-интенсивности соотв. падающего и прошедшего через в-во света, d-толщина слоя, k-коэф. поглощения, к-рый не зависит от толщины поглощающего слоя и интенсивности падающего излучения. Для характеристики поглощат. способности широко используют коэф. экстинкции, или светопоглощения; k' = k/2,303 (в см-1) и оптич. плотность А = lg I0/I, а также величину пропускания Т= I/I0. Отклонения от закона известны только для световых потоков чрезвычайно большой интенсивности (для лазерного излучения). Коэф. k зависит от длины волны падающего света, т.к. его величина определяется электронной конфигурацией молекул и атомов и вероятностями переходов между их электронными уровнями. Совокупность переходов создает спектр поглощения (абсорбции), характерный для данного в-ва.
2. Закон Бера: каждая молекула или атом независимо от относит. расположения др. молекул или атомов поглощает одну и ту же долю энергии излучения, т.е. , где с-концентрация в-ва. Если с выражена в моль/л,наз. молярным коэф. поглощения. Отклонения от этого закона свидетельствуют об образовании димеров, полимеров, ассоциатов, о хим. взаимодействии поглощающих частиц.
3. Объединенный закон Бугера-Ламберта-Бера:
Вид спектра поглощения определяется как природой образующих его атомов и молекул, так и агрегатным состоянием в-ва. Спектр разреженных атомарных газов - ряд узких дискретных линий, положение к-рых зависит от энергии основного и возбужденных электронных состояний атомов. Спектры молекулярных газов - полосы, образованные тесно расположенными линиями, соответствующими переходам между колебательным и вращательным энергетич. уровнями молекул. Спектр в-ва в конденсиров. фазе определяется не только природой составляющих его молекул, но и межмол. взаимодействиями, влияющими на структуру электронных уровней. Обычно такой спектр состоит из ряда широких полос разл. интенсивности. Иногда в нем проявляется структура колебат. уровней (особенно у кристаллов при охлаждении). Прозрачные среды, напр. вода, кварц, не имеют в спектре полос поглощения, а обладают лишь границей поглощения.
По спектрам поглощения проводят качеств. и количеств. анализ в-в. Абсорбционная спектроскопия широко применяют для изучения строения в-ва. Она особенно эффективна при исследовании процессов в жидких средах; по изменениям положения, интенсивности и формы полос поглощения судят об изменениях состава и строения поглощающих свет частиц без их выделения из р-ров.
В эмиссионных методах анализируемая проба в результате ее возбуждения излучает фотоны (кванты). Важнейшие эмиссионные методы - атомно-эмиссионный спектральный анализ (АЭС) и люминесцентный анализ.
Атомно-эмиссионная спектроскопия (АЭС) - спектроскопический метод анализа, основанный на измерении электромагнитного излучения оптического диапазона, испускаемого термически возбуждёнными свободными атомами или одноатомными ионами.
Атомно-эмиссионный спектральный анализ. Во всех вариантах АЭС пробу вносят в источник возбуждения, где тем или иным способом создается высокая температура (тысячи градусов). Образуется плазма (совокупность возбужденных атомов, ионов и электронов). В ней последовательно проходят следующие процессы: · испарение пробы; · атомизация первоначальных продуктов испарения (молекул или ионов); · возбуждение образовавшихся атомов; · испускание света возбужденными атомами.
Возникающее в ходе анализа полихроматическое излучение пробы фокусируют и направляют на входную щель спектрального прибора, где оно разлагается в спектр и регистрируется соответствующим приемником (фотоэлемент, диодная линейка, фотопластинка и др.). Можно наблюдать спектр и визуально (именно так работали Бунзен и Кирхгоф), однако это небезопасно для глаз. Для качественного анализа полученный спектр сопоставляют с эталонными спектрами разных элементов. По одному спектру пробы можно быстро и надежно обнаружить многие, а то и все присутствующие в ней элементы. Для количественного анализа надо измерить интенсивность излучения пробы на некоторых заранее выбранных длинах волн. На практике измеряют не саму интенсивность излучения (число квантов, испускаемых пробой в единицу времени), а зависящие от нее другие величины - фототок, абсолютное или относительное почернение фотопластинки и др. По этим аналитическим сигналам и рассчитывают содержания разных элементов, пользуясь градуировочными графиками, заранее полученными с помощью подходящих эталонов.
Приборы для спектрального анализа включают в себя три основных блока: блок возбуждения, диспергирующее устройство и блок регистрации излучения.
Источники возбуждения. В качестве источников возбуждения применяют пламя, электрическую дугу, искру, а также высокочастотную индуктивно-связанную плазму (ИСП, ICP).
Способы регистрации спектра. Фотографическая регистрация
Схема спектрографа. После разложения излучения пробы по длинам волн оно направляется на фотопластинку, содержащую в своем поверхностном слое кристаллы бромида серебра. В местах, куда попадет излучение, образуется металлическое серебро. В результате проявления и закрепления фотопластинки ее почернение во много раз усиливается. На пластинке остается спектр пробы в виде ряда черных линий одинаковой высоты и ширины, но с разной степенью почернения (картинка напоминает штрих-код товара). Все эти линии являются фотографиями входной щели, сделанными на разных длинах волн, соответственно спектральному составу излучения пробы. На одну и ту же фотопластинку можно последовательно сфотографировать десятки спектров, размещая их друг под другом.
Почернение аналитических линий на проявленной фотопластинке измеряют с помощью вспомогательного прибора – микрофотометра.
Фотоэлектрическая регистрация основана на применении фотоэффекта.
Количественный анализ. Интенсивность излучения в АЭС определяется концентрацией возбужденных атомов. Если все условия анализа одинаковы, получим прямо пропорциональную зависимость интенсивности излучения на данной длине волны от концентрации элемента в пробе:
Расчет концентраций ведут по предварительно построенным градуировочным графикам.
Люминесцентный анализ. Принцип метода и области его применения. В определенных условиях часть поглощенной веществом энергии может выделиться в виде вторичного излучения. Это явление называют люминесценцией. Кванты вторичного излучения, испускаемого люминесцирующими атомами, молекулами или ионами, имеют меньшую энергию, чем кванты, которые те же частицы поглощали при своем возбуждении. Виды люминесценции классифицируют по способу возбуждения. Наиболее известны: 1. Катодолюминесценция (свечение под действием потока электронов, например, свечение экрана кинескопа или жидкокристаллического экрана, свечение ламп дневного света). 2. Хемилюминесценция (свечение за счет химической реакции, например, у светлячков). 3. Фотолюминесценция. В этом случае проба светится за счет облучения невидимым УФ-светом от внешнего источника. Именно этот вид люминесценции обычно применяют в химическом анализе.
Рефрактометрия (от лат. refractus - преломленный и греч. metreo - измеряю) - это метод исследования веществ, основанный на определении показателя (коэффициента) преломления (рефракции) и некоторых его функций. Рефрактометрия (рефрактометрический метод) применяется для идентификации химических соединений, количественного и структурного анализа, определения физико-химических параметров веществ.
Показатель преломления n, представляет собой отношение скоростей света в граничащих средах. Для жидкостей и твердых тел n обычно определяют относительно воздуха, а для газов - относительно вакуума. Значения n зависят от длины волны l света и температуры, которые указывают соответственно в подстрочном и надстрочном индексах.
В случае газов необходимо также учитывать зависимость n от давления (указывать его или приводить данные к нормальному давлению).
В идеальных системах (образующихся без изменения объема и поляризуемости компонентов) зависимость показателя преломления от состава близка к линейной, если состав выражен в объемных долях (процентах) n=n1V1+n2V2 , где n, n1 ,n2 - показатели преломления смеси и компонентов, V1 и V2 - объемные доли компонентов (V1 + V2 = 1).
Для рефрактометрии растворов в широких диапазонах концентраций пользуются таблицами или эмпирическими формулами, важнейшие из которых (для растворов сахарозы, этанола и др.) утверждаются международными соглашениями и лежат в основе построения шкал специализированных рефрактометров для анализа промышленной и сельскохозяйственной продукции.
Зависимость показателя преломления водных растворов некоторых веществ от концентрации:
Влияние температуры на показатель преломления определяется двумя факторами: изменением количества частиц жидкости в единице объема и зависимостью поляризуемости молекул от температуры. Второй фактор становится существенным лишь при очень большом изменении температуры.
Температурный коэффициент показателя преломления пропорционален температурному коэффициенту плотности. Поскольку все жидкости при нагревании расширяются, то их показатели преломления уменьшаются при повышении температуры. Температурный коэффициент зависит от величины температуры жидкости, но в небольших температурных интервалах может считаться постоянным.
Обычно n жидких и твердых тел рефрактометрией определяют с точностью до 0,0001 на рефрактометрах, в которых измеряют предельные углы полного внутреннего отражения. Наиболее распространены рефрактометры Аббе с призменными блоками и компенсаторами дисперсии, позволяющие определять nD в "белом" свете по шкале или цифровому индикатору. Максимальная точность абсолютных измерений (10-10) достигается на гониометрах с помощью методов отклонения лучей призмой из исследуемого материала. Для измерения n газов наиболее удобны интерференционные методы. Интерферометры используют также для точного (до 10 -7) определения разностей n растворов. Для этой же цели служат дифференциальные рефрактометры, основанные на отклонении лучей системой двух-трех полых призм.
Автоматические рефрактометры для непрерывной регистрации n в потоках жидкостей используют на производствах при контроле технологических процессов и автоматическом управлении ими, а также в лабораториях для контроля ректификации и как универсальные детекторы жидкостных хроматографов.
Поляриметрия — методы исследования излучения, основанные на измерении: 1) степени поляризации излучения (света, радиоволн), 2) оптической активности веществ или их растворов
Поляриметрия используется для исследования излучений, а также в аналитической и структурной химии.
Теория поляриметрии Оптическая активность веществ очень чувствительна к изменениям пространственной структуры молекул и к межмолекулярному взаимодействию.
Поляризуемость атомов, ионов и молекул определяет степень межмолекулярного взаимодействия и его влияние на оптическую активность среды.
Поляриметрия даёт ценную информацию о природе заместителей в органических молекулах, о строении комплексных неорганических соединений.
Исследование оптической активности веществ С помощью оптических поляриметров определяют величину вращения плоскости поляризации света при прохождении его через оптически-активные среды (твёрдые вещества или растворы).
Поляриметрия широко применяется в аналитической химии для быстрого измерения концентрации оптически-активных веществ, для идентификации эфирных масел и в других исследованиях.
Величина оптического вращения в растворах зависит от их концентрации и специфических свойств оптически-активных веществ.
Измерение вращательной дисперсии света (спектрополяриметрия, определение угла вращения при изменении длины волны света позволяет изучать строение веществ.
Электрохимические методы анализа - совокупность методов качественного и количественного анализа, основанных на электрохимических явлениях, происходящих в исследуемой среде или на границе раздела фаз и связанных с изменением структуры, химического состава или концентрации анализируемого вещества. Э. м. а. делятся на пять основных групп: потенциометрию, вольтамперометрию, кулонометрию, кондуктометрию и диэлектрометрию.
Потенциометрия объединяет методы, основанные на измерении эдс обратимых электрохимических цепей, когда потенциал рабочего электрода близок к равновесному значению. Потенциометрия включает редоксметрию, ионометрию и потенциометрическое титрование.Вольтамперометрия основана на исследовании зависимости тока поляризации от напряжения, прикладываемого к электрохимической ячейке, когда потенциал рабочего электрода значительно отличается от равновесного значения. По разнообразию методов вольтамперометрия — самая многочисленная группа из всех Э. м. а., широко используемая для определения веществ в растворах и расплавах.Кулонометрия объединяет методы анализа, основанные на измерении количества вещества, выделяющегося на электроде в процессе электрохимической реакции в соответствии с Фарадея законами. При кулонометрии потенциал рабочего электрода отличается от равновесного значения. Различают потенциостатическую и гальваностатическую кулонометрию, причём последняя включает прямой и инверсионный методы, электроанализ и кулонометрическое титрование. К кондуктометрии относятся методы, в которых измеряют Электропроводность электролитов (водных и неводных растворов, коллоидных систем, расплавов, твёрдых веществ). Кондуктометрический анализ основан на изменении концентрации вещества или химического состава среды в межэлектродном пространстве; он не связан с потенциалом электрода, который обычно близок к равновесному значению. Кондуктометрия включает прямые методы анализа (используемые, например, в Солемерах) и косвенные (например, в газовом анализе) с применением постоянного или переменного тока (низкой и высокой частоты), а также хронокондуктометрию, низкочастотное и высокочастотное титрование.Диэлектрометрия объединяет методы анализа, основанные на измерении диэлектрической проницаемости вещества, обусловленной ориентацией в электрическом поле частиц (молекул, ионов), обладающих дипольным моментом. Методы диэлектрометрии применяют для контроля чистоты диэлектриков, например для определения малых количеств влаги. Диэлектрометрическое титрование используют для анализа растворов.
Газовая хроматография (ГХ) - хроматография, в которой подвижная фаза находится в состоянии газа или пара - инертный газ (газ-носитель). Неподвижной фазой является высокомолекулярная жидкость, закрепленная на пористый носитель или на стенки длинной капиллярной трубки, или только твердое пористое вещество, заполняющее колонку , в следствии чего газовая хроматография подразделяется на газо-жидкостную и газо-твердофазную. Газовая хроматография - универсальный метод разделения смесей разнообразных веществ, испаряющихся без разложения. При этом компоненты разделяемой смеси перемещаются по хроматографической колонке с потоком газа-носителя. По мере движения разделяемая смесь многократно распределяется между газом-носителем (подвижной фазой) и неподвижной фазой. Принцип разделения - неодинаковое сродство веществ к летучей подвижной фазе и стационарной фазе в колонке. Компоненты смеси селективно задерживаются последней, поскольку сродство их к этой фазе различно, и таким образом разделяются (компонентам с большим сродством требуется большее время для выхода из неподвижной фазы, чем компонентам с меньшим сродством). Затем вещества выходят из колонки и регистрируются детектором. Сигнал детектора записывается в виде хроматограммы автоматическим потенциометром (самописцем) или же регистрируется компьютером.
Области применения газовой хроматографии. Метод ГХ — один из самых современных методов многокомпонентного анализа, его отличительные черты — экспрессность, высокая точность, чувствительность, автоматизация. Метод позволяет решить многие аналитические проблемы. Количественный ГХ анализ можно рассматривать как самостоятельный аналитический метод, более эффективный при разделении веществ, относящихся к одному и тому же классу (углеводороды, органические кислоты, спирты и т.д.).
Жидкостная хроматография - вид хроматографии, в которой подвижной фазой (элюентом) служит жидкость. Неподвижной фазой может быть твердый сорбент, твердый носитель с нанесенной на его поверхность жидкостью или гель. Различают колоночную жидкостную хроматографию, в которой через колонку, заполненную неподвижной фазой, пропускают порцию разделяемой смеси в-в в потоке элюента (под давлением или под действием силы тяжести), и тонкослойную жидкостную хроматографию, в к-рой элюент перемещается под действием капиллярных сил по плоскому слою сорбента, нанесенного на стеклянную пластинку или металлическую фольгу.
В высокоэффективной жидкостной хроматографии (ВЭЖХ) используют колонки диаметром до 5 мм, плотно упакованные сорбентом с частицами малого размера (3-10 мкм); давление для прокачивания элюента до 3.107 Па (ее называют также хроматографией высокого давления). Варианты ВЭЖХ - микроколоночная хроматография на наполненных колонках малого диаметра и капиллярная хроматография на полых и наполненных сорбентом капиллярных колонках. К жидкостной хроматографии обычно относят также гидродинамическую хроматографию, где неподвижная фаза отсутствует.
По механизму удерживания разделяемых веществ неподвижной фазой Жидкостная хроматография делится на осадочную хроматографию, адсорбционную, распределительную, ионообменную хроматографию (в т. ч. ионную хроматографию), ион-парную, лигандообменную, эксклюзионную (ситовую) и аффинную хроматографию (биоспецифическую). Осадочная жидкостная хроматография основана на различной растворимости осадков, образующихся при взаимодействии компонентов анализируемой смеси с реагентом-осадителем. Адсорбционная жидкостная хроматография в зависимости от относительной полярности сорбента и элюента подразделяется на нормально-фазную и обращенно-фазную. В первом случае адсорбция веществ происходит на полярном сорбенте из неполярного элюента благодаря донорно-акцепторному взаимодействию или образованию водородных связей. Во втором - на поверхности гидрофобизир. сорбента из полярного элюента. В распределительной жидкостной хроматографии разделение основано на распределении веществ между двумя жидкими фазами: неподвижной, нанесенной на поверхность носителя, и подвижной элюентом. В зависимости от полярности жидких фаз возможны нормально-фазный и обращeнно-фазный варианты. К распределительной жидкостной хроматографии относится и экстракционная, в которой неподвижной фазой служит органический экстрагент, нанесенный на твердый носитель, а подвижной - водный раствор разделяемых соединений. В ионообменной жидкостной хроматографии разделение основано на различной способности разделяемых ионов к р-ции ионного обмена с фиксированными ионами сорбента, образующимися в результате диссоциации ионогенных групп последнего. В зависимости от знака заряда фиксир. ионов различают катиониты (закреплен анион) и аниониты (закреплен катион). Разделение ионов регулируют подбором оптим. значений рН элюента и его ионной силы. Вариант ионообменной жидкостной хроматографии - ионная хроматография, в которой разделенные анионы (катионы) детектируют в виде к-т (соотв. оснований) высокочувствительным кондуктометрическим детектором, а высокоэффективные колонки наполнены поверхностно-активным ионитом с небольшой емкостью. Ион-парную жидкостную хроматографию можно рассматривать как комбинацию адсорбционной и ионообменной; в качестве неподвижной фазы используют гидрофобизир. адсорбент, а подвижной - водно-органический элюент с добавлением поверхностно-активных ионогенных соединений (ион-парных реагентов), напр. додецилсульфата Na или триметилцетиламмоний бромида. Разделение основано на удерживании ион-парного реагента на гидрофобной поверхности адсорбента с образованием ионита, который и проводит разделение ионогенных соединений. Возможно также образование ионных пар разделяемых ионов с ион-парным реагентом, которые затем удерживаются на гидрофобизир. поверхности адсорбента. Лигандообменная жидкостная хроматография основана на различной способности разделяемых соединений образовывать комплексы с катионами переходных металлов - Cu(II), Ni(II), Zn(II), Cd(II), Co(II) и др. - и фиксированными группами (лигандами) неподвижной фазы. Часть координац. сферы ионов металла занята молекулами воды или др. слабыми лигандами, которые могут вытесняться молекулами разделяемых соединений. Наиболее эффективна для разделения оптических изомеров. Аффинная жидкостная хроматография основана на образовании прочной связи со специфическими группами неподвижной фазы (лигандами, аффинантами). Взаимодействие лигандов с разделяемыми веществами основано на биол. ф-ции последних. Так, при разделении ферментов лигандами служат их субстраты, ингибиторы или коферменты, токсинов - рецепторы, белков - антитела и т. д. Особенно эффективна в биотехнологии и биомедицине для выделения ферментов, белков, гормонов. В эксклюзионной жидкостной хроматографии разделение основано на различиях в размерах молекул; молекулы малых размеров проникают в сравнительно тонкие поры сорбента и задерживаются в них, крупные молекулы либо не проникают в поры, либо проникают лишь в широкие поры и проходят колонку с незначительным удерживанием. Поверхность сорбента и состав элюента подбирают так, чтобы исключить или уменьшить энергию адсорбц. взаимодействия (однако иногда при разделении олигомеров удобнее использовать адсорбц. механизм). Применяют для разделения олигомеров и полимеров (в т. ч. биологических).
Электрофорез – метод разделения электрически заряженных частиц в растворе путем пропускания через этот раствор электрического тока. Направленное движение коллоидных частиц под действием внешнего электрического поля.
Скорость движения частиц зависит от их заряда, они разделяются, отходя к различным полюсам электрода
Капиллярный гель-электрофорез – проводится в капилляре заполненном гелем. На движение молекул оказывает влияние матрица геля, поэтому происходит селективное разделение молекул по размерам.
Область применения: изучение ДНК, РНК, белков
МАСС-СПЕКТРОМЕТРИЯ основана на прямом измерении отношения массы к числу элементарных положительных или отрицательных зарядов ионов (m/z), полученных из анализируемого вещества и находящихся в газовой фазе. Данное отношение выражается в атомных единицах массы (1 а.е.м. = одной двенадцатой массы нуклида 12С) или в дальтонах (1 Да = массе атома водорода).
Ионы, образовавшиеся в ионном источнике, ускоряются и перед попаданием в детектор разделяются с помощью масс-анализатора. Все эти действия происходят в камере, в которой насосная система поддерживает вакуум от 10-3 до 10-6Па.
Результирующий масс-спектр является графиком зависимости относительного количества различных ионов от отношения m/z. Сигнал, отвечающий иону, представлен несколькими пиками, соответствующими статистическому распределению различных изотопов этого иона. Такой сигнал называется изотопным профилем, а пик (по крайней мере, для небольших молекул), представляющий наиболее распространённые изотопы для каждого атома, - моноизотопным пиком.
Масс-спектрометрический анализ даёт важную качественную (определение молекулярных масс; информация, касающаяся структуры определяемых фрагментов) и количественную (с использованием внешнего или внутреннего стандартов) информацию с пределом обнаружения от пикомоля до фемтомоля.
ВВОД ОБРАЗЦА → ионизируется → проходит через поток электронов
Первой стадией анализа является ввод образца в прибор без значительного нарушения вакуума. В широко распространённом методе, называемом прямым вводом жидкости, образец помещается на конец цилиндрического штока (в кварцевом тигле, на проволоке или на металлической поверхности). Этот шток через вакуумный шлюз, в котором поддерживается первичный промежуточный вакуум между атмосферным давлением и вторичным вакуумом прибора, вводится в спектрометр.
Другие системы ввода позволяют проанализировать компоненты смеси, так как они разделяются с помощью соответствующего прибора, соединённого с масс-спектрометром.
Газовая хроматография/масс-спектрометрия. При использовании подходящих колонок (капиллярных или полукапиллярных) возможно непосредственное введение конца колонки в ионный источник прибора без применения сепаратора.
Жидкостная хроматография/масс-спектрометрия. Такая комбинация особенно полезна для анализа полярных соединений, которые являются недостаточно летучими либо слишком термолабильными, для того чтобы их можно было проанализировать методом газовой хроматографии в сочетании с масс-спектрометрией. Данный метод осложняется трудностью получения ионов в газовой фазе из жидкой фазы, что требует применения специальных интерфейсов, таких как:
- прямой жидкостный ввод: подвижная фаза распыляется и растворитель испаряется перед ионным источником прибора, (используется испаритель)
- интерфейс с пучком частиц: подвижная фаза, скорость которой может достигать 0,6 мл/мин, распыляется в десольватационной камере, в результате в ионный источник прибора попадают лишь анализируемые вещества в нейтральной форме; данный способ может быть использован для соединений с относительно низкой полярностью и молекулярными массами меньше 1000 Да,
- интерфейс с движущейся полосой: подвижная фаза, скорость которой может достигать 1 мл/мин, наносится на поверхность движущейся полосы; после выпаривания растворителя анализируемые вещества переносятся к ионному источнику прибора, где подвергаются ионизации; данный способ плохо подходит для очень полярных или термолабильных соединений.
Другие виды интерфейсов (электрораспыление, термораспыление, химическая ионизация при атмосферном давлении) могут рассматриваться как самостоятельные методы ионизации и описаны в разделе, посвященном способам ионизации.
Сверхкритическая флюидная хроматография/масс-спектрометрия.
Подвижная фаза, обычно состоящая из находящегося в сверхкритическом состоянии диоксида углерода, переходит в газообразное состояние после прохождения через нагретую заслонку, находящуюся между колонкой и ионным источником.
Капиллярный электрофорез/масс-спектрометрия, Элюент вводится в ионный источник, в некоторых случаях после добавления дополнительного растворителя, для того чтобы достичь скорости потока порядка нескольких миллилитров в минуту. Ограничениями данного метода являются малые количества вводимого образца и необходимость использовать летучие буферные растворы.
ТЕРМИЧЕСКИЙ АНАЛИЗ, метод исследования физ.-хим. и хим. процессов, основанный на регистрации тепловых эффектов, сопровождающих превращения в-в в условиях программирования т-ры. Поскольку изменение энтальпии DH происходит в результате большинства физ.-хим. процессов и хим. р-ций, теоретически метод применим к очень большому числу систем. Установка для термического анализа включает печь, держатели для образцов, термопары (с самописцами), измеряющими т-ру печи и образцов. Для записи кривых в координатах т-ра-время используют фоторегистрирующие пирометры и автоматич. потенциометры.
В термическом анализе можно фиксировать т. наз. кривые нагревания (или охлаждения) исследуемого образца, т.е. изменение т-ры последнего во времени. В случае к.-л. фазового превращения в в-ве (или смеси в-в) на кривой появляются площадка или изломы. Большей чувствительностью обладает метод дифференциального термического анализа (ДТА), в к-ром регистрируют во времени изменение разности т-р DT между исследуемым образцом и образцом сравнения (чаще всего Аl2О3), не претерпевающим в данном интервале т-р никаких превращений. Минимумы на кривой ДТА (см., напр., рис.) соответствуют эндотермич. процессам, а максимумы - экзотермическим. Эффекты, регистрируемые в ДТА, м. б. обусловлены плавлением, изменением кристаллич. структуры, разрушением кристаллич. решетки, испарением, кипением, возгонкой, а также хим. процессами (диссоциация, разложение, дегидратация, окисление-восстановление и др.). Большинство превращений сопровождается эндотермич. эффектами; экзотермичны лишь нек-рые процессы окисления-восстановления и структурного превращения. На вид кривых ДТА, как и на вид кривых в термогравиметрии, оказывают влияние мн. факторы, поэтому воспроизводимость метода, как правило, плохая.
Обычно данные ДТА используют в сочетании с результатами термогравиметрич., масс-спектрометрич. и дилато-метрич. исследований (см. Дериватография). Это позволяет, напр., делать выводы об обратимости фазовых превращений, изучать явления переохлаждения, образование мета-стабильных фаз (в т. ч. короткоживущих). Мат. соотношения между площадью пика на кривой ДТА и параметрами прибора и образца позволяют определять теплоту превращения, энергию активации фазового перехода, нек-рые кинетич. константы, проводить полуколичеств. анализ смесей (если известны DH соответствующих р-ций). С помощью ДТА изучают разложение карбоксилатов металлов, разл. металлоорг. соединений, оксидных высокотемпературных сверхпроводников. Этим методом определили температурную область конверсии СО в СО2 (при дожигании автомобильных выхлопных газов, выбросов из труб ТЭЦ и т.д.)
Выборкой называется последовательность независимых одинаково распределённых случайных величин. Выборка, пронумерованная в порядке возрастания, т.е. x1, x2 ... xn, называется вариационным рядом. Сами значения x называются вариантами, а n - объёмом выборки. В табл. 10.1 приведены основные характеристики, используемые для описания выборки.
Чем меньше число степеней свободы (n-1), тем в большей степени выборочные характеристики отличаются от характеристик случайной величины. Для характеристики выборок малых объёмов (n < 30), взятых из нормально распределённых генеральных совокупностей, используют распределение Стьюдента (t-распределение), представляющее собой распределение случайной величины t
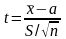 (или
(или
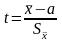 )
)
Данное
распределение зависит только от объёма
выборки и не зависит от неизвестных
параметров a
и .
При
 распределение Стьюдента переходит в
стандартное нормальное распределение.
распределение Стьюдента переходит в
стандартное нормальное распределение.
Распределение Стьюдента можно использовать для расчёта доверительного интервала выборочного среднего (в том случае, если выборка имеет нормальное распределение). Доверительным интервалом называется интервал, вероятность попадания значений случайной величины в который равна принятой нами доверительной вероятности 1-, где - уровень значимости (в аналитической практике = 0,05). Неизвестное математическое ожидание с вероятностью 1- попадёт в интервал:
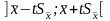
Например,
если
= 0,05 и f
= 5, то доверительный интервал для
выборочного среднего равен 2,57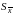 .
.
10.6. Пример статистической обработки результатов измерений. Исключение промахов
Процесс анализа многостадиен. Каждая стадия вносит определённый вклад в неопределённость окончательного результата. Рассмотрим простейший вариант статистической обработки последней стадии анализа - измерения аналитического сигнала.
Пример 10.4. При измерении рН раствора с помощью рН-метра получены следующие результаты 4,32; 4,35; 4,36; 4,98; 4,38; 4,34. Провести статистическую обработку полученных результатов.
Перед началом статистической обработки необходимо проверить, не содержат ли полученные результаты грубых погрешностей. Измерения, в которых обнаружены такие погрешности, должны быть исключены. Их нельзя использовать при дальнейшей статистической обработке результатов. Существует несколько способов исключения грубых погрешностей. Для исключения промахов при работе с выборками малого объёма (n = 4 - 10) можно воспользоваться величиной Q-критерия. Для выборок больших объёмов можно использовать, например, «правило 3» - если значение отличается от среднего более, чем на 3 стандартных отклонения, то его можно считать промахом.
Экспериментальное значение Q-критерия рассчитывают по следующим формулам:

Полученное значение сравнивают с критической (табличной) величиной для Q-критерия. Если оно превышает последнюю, то проверяемый результат является промахом и его необходимо исключить из дальнейших расчётов.
Преобразуем выборку, приведенную в примере 10.4, в вариационный ряд:

Последнее значение является явно подозрительным. Рассчитаем для него величину Q
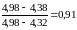
Для n= 6 и P = 0,90 Qкрит = 0,48. Следовательно, результат рН = 4,98 является промахом и его необходимо исключить.
При обработке
оставшихся данных с помощью формул,
представленных в табл. 10.1, получены
следующие результаты:
 =
4,35;
=
4,35;
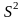 = 5,0010-4;
= 5,0010-4;
 = 2,2410-2;
= 2,2410-2; = 1,0010-2;
= 1,0010-2;
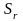 = 5,1510-3;
= 5,1510-3;
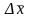 (=0,05)
= 0,03.
Таким образом, рН
= 4,350,03.
(=0,05)
= 0,03.
Таким образом, рН
= 4,350,03.
Обратите внимание, что окончательный результат среднего значения рН содержит столько же значащих цифр (3), сколько их присутствует в исходных данных. Величина, характеризующая доверительный интервал среднего, имеет столько же десятичных знаков (2), сколько и само среднее. Если бы мы привели в качестве результата, что-нибудь вроде 4,35000,028, то это было бы неверно.
10.7. Основные характеристики методики анализа
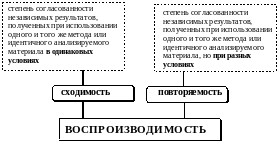
Основными характеристиками методики анализа являются воспроизводимость и правильность, предел обнаружения, границы определяемых содержаний и чувствительность.
Воспроизводимость
Воспроизводимость (precision) - степень близости друг к другу независимых результатов измерений при оговоренных условиях.
Количественно воспроизводимость (или невоспроизводимость) удобнее всего описывать с помощью относительного стандартного отклонения. Чем больше величина Sr, тем воспроизводимость хуже.
Для сравнения воспроизводимости результатов двух серий анализа используют F-критерий (критерий Фишера).

Полученное значение сравнивают с критическим (табличным) при выбранном уровне значимости (обычно 0,05 или 0,01) и числе степеней свободы (f1,f2). При конкурирующей гипотезе «одна из дисперсий больше второй дисперсии» используют уровень значимости (односторонняя постановка задачи), при конкурирующей гипотезе «дисперсии не равны между собой» используется уровень значимости /2 (двухсторонняя постановка задачи). Если Fэксп < Fкрит, то считается, что дисперсии двух серий анализа отличаются незначимо.
Пример 10.5.
При
измерении рН раствора один студент
получил результат
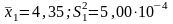 ,
а второй студент -
,
а второй студент -
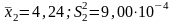 .
Определить, различаются ли полученные
данные по воспроизводимости, если каждый
студент провёл по 5 параллельных
измерений.
.
Определить, различаются ли полученные
данные по воспроизводимости, если каждый
студент провёл по 5 параллельных
измерений.

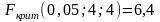
Поскольку
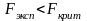 ,
можно сделать вывод, что полученные
результаты имеют одинаковую
воспроизводимость.
,
можно сделать вывод, что полученные
результаты имеют одинаковую
воспроизводимость.