

6 курс / Неонатология / Пороки_развития_и_наследственные_заболевания_нервной_системы_у_детей
.pdf(псевдопапиледема). С рождения или спустя 1-3 года на коже и в подкожной
клетчатке появляются гемангиомы.
1)Найдите соответствие между заболеваниями и его признаками:
1.Синдром Штурге-Вебера
2.Односторонняя макроэнцефалия или частичная макроэнцефалия
a)“пламенный невус” (на лице и голове, почти всегда односторонний, в зоне иннервации тройничного нерва, чаще I ветвь); эпилепсия (парциальные припадки - на контралатеральной стороне), врожденная глаукома с одностор.гидрофтальмией (не всегда).Также характерно слабоумие.
b)выражается в увеличении только одного полушария головного мозга.
асимметрия черепа, выявляемая с момента рождения, сопровождается в
клинической практике судорожными состояниями.
2) Выберети правильные ответы. Укажите первичные признаки туберозного
склероза:
a)классические шагреновые пятна, ретинальная гамартрома
b)лицевой ангиофиброматоз
c)субэпиндемальные глиальные узелки, почечный ангиолипоматоз
d)«младенческие» судороги
e)рабдомиома сердца
Ситуационная задача: Ребенок 2 года. Множественные гемангиомы на передней и задней поверхности тела, размер черепа увеличен, невростатус без особенностей. На глазном дне отек соска зрительного нерва. На нейросонографии признаков внутричерепной гипертензии нет. Укажите ваш предварительный диагноз:
ГЛАВА 13
Болезни, связанные первичным поражением серого вещества головного мозга:
GM -1 ганглиозидоз, GM-2 ганглиозидоз - болезнь Тея-Сакса, болезнь Сандхофа,
болезнь Нимана-Пика, болезнь Гоше, болезнь Альперса
231
Учебная цель: В процессе обучения магистрант должен изучать клинику,
диагностику, терапию и прогноз GM -1- ганглиозидоза, GM-2 ганглиозидоз-болезни Тея-Сакса, болезни Сандхофа, болезни Нимана-Пика (сфингомиелиновый липидоз),
болезни Гоше (глюкоцереброзидоз); болезни Альперса (прогрессирующая полиодистрофия мозга)
Магистрант должен знать:
1. Уметь дифференцировать GM -1- ганглиозидоз, GM-2 ганглиозидоз-болезнь Тея-Сакса, болезнь Сандхофа, болезнь Нимана-Пика, (сфингомиелиновый липидоз), болезнь Гоше (глюкоцереброзидоз); болезнь Альперса
(прогрессирующая полиодистрофия мозга).
2.Характерные признаки GM -1- ганглиозидоза, GM-2 ганглиозидоз-болезни Тея-
Сакса, болезни Сандхофа, болезни Нимана-Пика (сфингомиелиновый липидоз),
болезни Гоше (глюкоцереброзидоз); болезни Альперса (прогрессирующая полиодистрофия мозга)
3. Клинические проявления GM -1- ганглиозидоза, GM-2 ганглиозидоз-болезни Тея-Сакса, болезни Сандхофа, болезни Нимана-Пика (сфингомиелиновый липидоз), болезни Гоше (глюкоцереброзидоз); болезни Альперса
(прогрессирующая полиодистрофия мозга)
4.Диагностику, лечение и прогноз GM -1- ганглиозидоза, GM-2 ганглиозидоз-
болезни Тея-Сакса, болезни Сандхофа, болезни Нимана-Пика
(сфингомиелиновый липидоз), болезни Гоше (глюкоцереброзидоз); болезни Альперса (прогрессирующая полиодистрофия мозга)
Ганглиозидозы - заболевания характеризующиеся локализацией липидных включений, которые преимущественно расположены в ядрах (ганглиях0 серого вещества, и лишь незначительное их количество находится в миелине.
Патогенез. Ганглиозиды являются компонентами клеточных плазматических мемабран и состоят из сфингозина, жирных кислот, гексозы, гексозамина и нейраминовой кислоты. Существует много вариантов ганглиозидов, которые сменяют
232
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
друг друга в зависимости от возраста и состояния организма. Сейчас известно 10
видов ганглиозидов, 4 из них составляют 90% отобщей фракции ганглиозидов мозга.
Их функции важны и разнообразны: формирование мембранных рецепторов для ферментов, участие в процессах клеточной адгезии, образование сигнальных комплексов для нейротрансмиттеров, образование связей между нейронами.
Нарушение всех этих функций и лежит в основе патогенеза ганглиозидов. В норме старые ганглиозиды при обновлении мембран разрушаются гидролитическими ферментами внутри лизосом до образования сфингозина и жирных кислот. При дефекте расщепляющего их ферментагексаминидазы ганглиозиды не подвергаются распаду, а депонируются в цитоплазме нейронов. Поскольку ганглиозиды распространены не только в ЦНС, но и в некоторых внутренних органах (печень и селезенка), развивается также гепатоспленомегалия.
Патологическая анатомия. Макроскопически отмечается увеличение мозга и наряду с этим диффузная атрофия отдельных областей, чаще мозжечка, затылочных долей.
Выявляется истончение зрительных нервов. Гистологически обнаруживается картина генерализованного распада ганглиозных клеток нервной системы, особенно клеток коры большого мозга и мозжечка. Дегенерация нервных клеток проявляется увеличением их размера и характерным набуханием, «баллонообразным» вздутием клеток и их отростков. Практически каждый нейрон коры изменен, его ядро смещено к периферии, а цитоплазма заполнена жирорастворимым веществом. по мере прогрессирования болезни число живых нейронов уменьшается и становиться больше клеток-тканей. Нейрофибриллы и тигроидное вещество распадаются полностью.
Задерживается миелинизация белого вещества, а втерминальных стадиях происходит его рапад (демиелинизация) с образованием множественных мелких кист, и белое вещество приобретает спонгиозный вид. Те же изменения можно видеть в грушевидных клетках мозжечка (клетки Пуркинье) и крупных нейронах ствола и спинного мозга. Ганглиозные клетки сетчатки , особенно в области желтого пятна,
раздуты от липидных включений. Чем больше включений в нейроне, тем больше в размере сам нейрон, постепенно его отростки деформируются и исчезают шипики дендритов, а внутриклеточные органеллы подвергаются механическому разрушению.
233
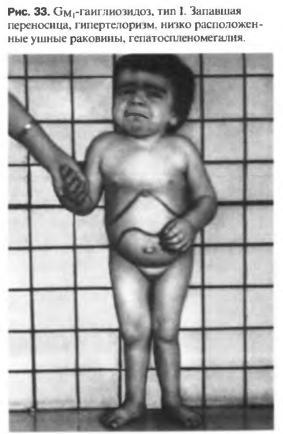
Формируются патологические синапсы. ГАМКергические контакты уступают место холинергическим. Патоморфологические изменения настолько характерны, что позволяют установить диагноз.
GM-1 ганглиозидоз 1го типа
МКБ-10 • E75.1 Другие ганглиозидозы
Синонимы : GM1-GANGLIOSIDOSIS, TYPE I, Семейный нейровисцеральный липидоз; недостаточность В-галактозидазы
Клиника : Дети с данным заболеванием резко отстают в психомоторном развитии с раннего возраста. У больных отмечаются плоская переносица, гипертелоризм, выступающий лоб,
низко расположенные уши, гипертрофия десен.
После 6 мес. выявляется гепатомегалия,
спленомегалия выражена меньше. Со стороны скелетной системы обнаруживают брахидактилию, кифосколиоз, множественные сгибательные контрактуры суставов;
рентгенологически находят признаки дизостоза.
При офтальмологическом исследовании у 50%
больных выявляют «вишнёвую косточку». В
начале 2-го года жизни появляются затруднения при глотании , тонико-клонические судороги, развиваются амавроз, отсутствие
реакции на окружающее. Дети погибают в возрасте 2-3 лет от сопутствующих бронхолёгочных инфекций. При гистологическом исследовании выявляется вакуолизация лимфоцитов, клеток костного мозга, гепатоцитов, клеток почечного эпителия, кожных фибробластов. Гистохимическими методами в этих клетках обнаруживают кислые мукополисахариды, ганглиозиды, липиды. Основной дефект заключается в недостаточности лизосомального фермента В-галактозидазы.
234
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
Минимальные диагностические признаки :
Висцеромегалия, характерные изменения лица и костей; вакуолизация клеток костного мозга и лмфоцитов; нарастание GM1-ганглиозида в клетках головного мозга и ганглиозидов и мукополисахаридов во всех органах и тканях; дефицит В-
галактозидазы в мозге, печени, культуре фибробластов,лейкоцитах и моче.
Течение, Прогноз :
Популяционная частота неизвестна.
Соотношение полов – М1:Ж1
Тип наследования-аутосомно-рецессивный
Дифференциальный диагноз :
Мукополисахаридозы; GM2-ганглиозидоз с дефицитом гексозаминидазы А;
Нимана=Пика болезнь; GM1 – ганглиозидоз, тип II; фукозидоз; муколипидозы.
GM-1 ганглиозидоз 2го типа
МКБ-10 • E75.1 Другие ганглиозидозы
Синонимы : GM1-GANGLIOSIDOSIS, TYPE II, Ювенильный системный липидоз;
болезнь Дерри
Клиника :
Типичны отставание в психомоторном развитии (с 6-12 мес), прогрессирующий спастический тетрапарез, атаксия, мышечная гипотония, сменяющаяся ригидностью,
судорогами, деменцией. терминальная стадия характеризуется децеребрацией.
продолжительность жизни – от3х до 10 лет. Рентгенологически выявляется дизостоз (
в некоторых случаях) , но значительно менее выраженный по сравнению с I типом
GM1 – ганглиозидоза. «Пенистые» клетки обнаруживают в костном мозге.
Вакуолизированные клетки обнаруживаются в печени, селезенке и почечных клубочках. Происходит накопление GM1 ганглиозида в мозге и кератан-сульфат подобного полисахарида в печени. Отмечается дефицит В-галактозидазы в мозге,
печени, лейкоцитах и культуре фибробластов. Иногда незначительно повышены мукополисахариды в моче.
Минимальные диагностические признаки :
235
Спастический тетрапарез, атаксия, постепенно развивающееся отсутствие реакции на окружающее; обнаружение «пенистых» клеток в костном мозге; накопление GM1 –
ганглиозида в головном мозге; дефицит В-галактозидазы в тканях , лейкоцитах и моче.
Течение, Прогноз :
Популяционная частота неизвестна.
Соотношение полов –M1:Ж1.
Тип наследования – аутосомно-рецессивный
Дифференциальный диагноз :
GM1-ганглиозидоз тип I; цероидлипофусциноз неврональный; ювенильный липидоз,
другие сфинголипидозы.
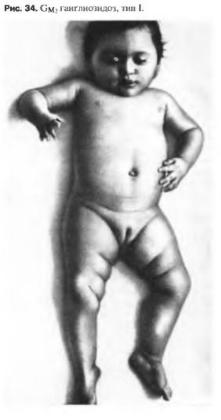
GM-2 ганглиозидоз 1го типа-наследственное заболевание, характеризующееся прогрессирующим снижением зрения и распадом интеллекта в сочетании с другими неврологическими симптомами.
МКБ-10 • E75.0 Ганглиозидоз-GM2
Синонимы :
GM1-GANGLIOSIDOSIS, TYPE I, GM-2 ганглиозидоз c дефицитом гексаминозидазы А, амавротическая идиотия Тея-Сакса
236
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
Этиология : Наследственность. Семейнфй характер болезни проявляется возникновением амавротической идиотии у братьев и сестер; родители остаются здоровыми. Заболевание передается по аутосомно-рецессивному типу с высокой пенетрантностью гена. Отмечается более частое развитие амавротической идиотии в популяции евреев восточноевропейской ветви.
Накопление ганглиозидов происходит в результате дефекта гексаминидазы. Её ген расположен на хромосоме 5 (5ql 1.2-13.3). Существует огромное количество (около80) мутаций этого гена, что обуславливает разнообразие клинических форм ганглиозидозов
Клиника : Ранняя детская форма Тея-Сакса часто носит семейный характер и начинается в возрасте 4-6 мес.
Ребенок, ранее активный, постепенно утрачивает статические и двигательные функции, теряет интерес к окружающему, перестает играть, Рано обнаруживается снижение зрения. Ребенок не может фиксировать взгляд на окружающих, не смотрит на блестящие предметы,
довольно быстро развивается слепота. Одновременно наблюдается снижение интеллекта до степени идиотии. Нередко развивается глухота.
Двигательные нарушения приводят к полной обездвиженности. При болезни Тея-
Сакса отмечается симптом повышенной реакции на звуковые раздражители и прикосновение-сильные вздрагивания. Часто бывают судороги, преимущественно тонического характера. Из других клинических симптомов обнаруживаются шейные тонические рефлексы Магнуса-Клейна, псевдобульбарные расстройства, вегетативно-
эндокринные симптомы, гипотермия, гипергидроз, отеки, оирение и др. Эти признаки часто варьируют. Болезнь Тея-Сакса длится в среднем1,5 -2 года, приводит к тяжелому истощению, полной обездвиженности. В конечной стадии болезни развивается клиническая картина децеребрационной ригидности. Дети умирают к концу 2-го года болезни.
237
Минимальные диагностические признаки : Задержка психомоторного развития;
симптом «вишнёвой косточки» на глазном дне; дефицит гексоаминозидазы А в сыворотке и тканях
Течение, Прогноз : Популяционная частота неизвестна.
Соотношение полов – M1:Ж1
Тип наследования- аутосомно-рецессивный. Ген локализован на 15q 23 q 24.
Популяционная частота-1:5000 среди евреев-ашкенази родом из Польши и Литвы.
Диагностика : В развернутой стадии болезни в области макулы появляется сероватобелый слегка проминирующий круглый очаг размером в 1,5-2 диаметра диска зрительного нерва. Цвет этого очага обусловлен генерализованным распадом и набуханием ганглиозных клеток сетчатки и их отростков. Истончение сетчатки дегенеративного характера в центре макулы приводит к образованию ярко-розового пятна (просвецивающая сосудистая оболочка). Изменения в области желтого пятна наиболее выражены, вплоть до атрофии зрительных нервов. Наличие характерных изменений на глазном дне (синдром вишневой косточки) позволяет считать диагноз болезни Тея-Сакса несомненным.
В цереброспинальной жидкости (ЦСЖ) повышен уровень сфинголипидов, отмечено увеличение общих фосфолипидов, главным образом кефалинов.
Продукты нарушенного обмена откладываются в сером веществе мозга и мозжечка,
причем их концентрация превышает нормальный уровень в 100-300 раз . В белом веществе мозга, печени, селезенке и периферической крови также повышено содержание ганглиозидов.
Важным признаком является наличие в периферической крови больных вакуолизированных лимфоцитов. При гистохимическом исследовании в вакуолях обнаруживаются липиды. На КТ уже на ранних стадиях болезни обнаруживаются гиподенситивныфе очаги и в хвостатом ядре, таламусе и скорлупе, на МРТ эти участки выглядят гиперинтенсивными.
Пренатальная диагностика основана на определении уровня гексаминидазы в амниотической жидкости или культуре фибробластов, но возможна только в III
триместре беременности. Оценка активности гексоминидазы в плазме крови является
238
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/
массовым скринингом, проводится взрослым людям, при планировании семьи,
относящимся к этническим группам с наибольшим распротранением заболевания. В
США и странах Европы это исследование является обязательным для восточно-
европейской популяции евреев.
Дифференциальный диагноз : Цероидлипофусциноз; GM1 – ганглиозидоз и лейкодистрофия; GM2-ганглиозидоз, тпи II
Лечение : Специфического лечения не разработано. Поскольку епонирование ганглиозидов в клетках мозга начинается уже во II триместре пренатального развития,
заместительная ферментотерапия и трансплантация костного мозга не оказывают выраженного положительного эффекта.
GM-2 ганглиозидоз 2го типа
МКБ-10 • E75.0 Ганглиозидоз-GM2
Синонимы : GM2-GANGLIOSIDOSIS, TYPE II, GM2-ганглиозидоз с дефицитом гексоаминидазы А и В; амавротическая идиотия Сандхоффа
Клиника : Типичны отставание в психомоторном развитии в первые 6 мес жизни,
гипотония. На глазном дне определяется симптом «вишнёвой косточки» . Характерны гепатоспленомегалия, «кукольное лицо», может быть микроцефалия. По мере прогрессирования развиваются миоклонические судороги , спастический тетрапарез.
Смерть наступает в 2-3 гола. В костном мозге обнаруживается « пенистые» клетки. В
основе заболевания лежит дефицит ферментов гексоаминидазы А и В, что приводит к накоплению ганглиозида GM2 в нервной системе и внутренних органах.
Минимальные диагностические признаки : Отставание в психомоторном развитии;
симптом «вишнёвой косточки» на глазном дне; дефицит гексоаминидазы А и В.
Течение, Прогноз : Популяционная частота неизвестна.
Соотношение полов – М1:Ж1
Тип наследования – аутосомно-рецессивный
Дифференциальный диагноз : GM2 – ганглиозидоз, тип I
Нимана-Пика болезнь. Это наследственное заболевание обмена
239
сфингофосфолипидов, при ктором происходит накопление сфингомиелина в лизосомах мозга, печени, ретикуло-эндотелиальной системе
Синонимы : Niemann-Pick disease, сфингомиелолипидоз
Этиология , Патогенез: Болезнь Нимана-Пика связана с дефицитом активности сфингомиелиназы и накоплением сфингомиелина в клетках ретикулоэнлотелиальной системы. Ген кислой сфингомиелиназы находится на хромосоме 11. Типы А и В болезни Нимана-Пика – аутосомно –рецессивные лизосомальные нарушения, их гены расположены на аллельных участках хромосомы 11. Ген клонирован, описано большое количество мутаций. Ген типа С болезни Нимана-Пика картирован, он находится на хромосоме 18 и кодирует белок, который переносит через цитоплазматическую мембрану холестерин и гликолипиды, поэтому при типе СЧ накапливается не только сфингомиелин, но и холестерин. В норме сфингомиелин содержиться в мозге, печени, почках и селезенке.
Паталогоанатомическое исследование обнаруживает значительное увеличение размеров и желтую окраску печени и селезенки, пятнистый рисунок легких.
Надпочечники также значительно увеличены в размерах и содержат большое количество липидов. При микроскопии во всех органах обнаруживаются клетки,
которые при фиксации спиртом выглядит «пенистыми» - клетки Нимана-Пика. Эти клетки могут достигать значительной величины 20-25мкм, в некоторых случаях 90
мкм. Пенистость клетокэто артефакт, вызванный рстворением жироподобных субстанций, содержащихся в клетках. Клетки Нимана-Пика могут быть онаружены при жизни в пунктатах селезнки, костного мозга. Крупные клетки с пенистой протоплазмойне специфичны для болезни Нимана-Пика и могут обнаруживаться при других липидозах и гиперлипидемии. Однако они хорошо отличаются от клеток Гоше.
Клиника : В начале болезни наблюдается отказ ребенка отпищи, периодическая рвота;
очень рано увеличиваются размеры печени и селезенки , развивается гипотрофия.
Появление признаов, указывающих на поражение нервной системы, также позволяет заподозрить болезнь Нимана-Пика. Спастические парезы могут сменяться общей мышечной гипотонией, гипорефлексией. Прогрессирующее поражение нервной системы ведет к резкому отставанию ребенка в нервно-психическом развитии,
240
Рекомендовано к покупке и прочтению разделом по неврологии сайта https://meduniver.com/