

Лекции_2 / ТЛ1_Основные понятия. СТП
.pdf_
Thin films of BaO-2TiO2 composition, deposited on the quartz substrate by means of CVD technique
. Выбираются наиболее перспективные стартовые реагентыt. |
|
. Синтезируются новые наноматериалы. |
21 |
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
11 |

_
•Экранирование центрального атома и методы его количественной оценки
•Изучение стерических факторов, а также разработка подходов к эмпирическим и качественным объяснениям пространственного экранирования центрального атома (ц.а.) позволяет посредством определения степени экранирования ц.а., расчета числа и определения типов ММК получить достаточно надежную
информацию о термохимических свойствах соединения, не
прибегая к их непосредственному экспериментальному исследованию. В случае, когда прямое экспериментальное определение данных параметров невозможно – это приобретает первостепенное значение.
•К настоящему времени, сформировались и активно развиваются ряд полуэмпирических подходов к описанию и определению экранирования центрального атома, числа и типов межмолекулярных контактов, опирающиеся на принцип аддитивности – это методы «телесных углов», «расширяющийся координационной сферы», «полиэдров Воронова-Дирихле» и некоторые другие. Исторически первым был метод «телесных углов». Метод «полиэдров Воронова-Дирихле» появился значительно позже и получил распространение в среде кристаллохимиков, так как оперирует с кристаллохимическими понятиями. Все методы реализованы в виде различных компьютерных программ.
• |
Метод «Телесных углов» |
|
|
• |
С.А.Толман |
попытался |
количественно |
|
охарактеризовать стерические факторы, используя |
||
|
понятие о телесном угле, описывая фосфиновые |
||
|
лиганды, и связал физические, химические и |
||
|
каталитические свойства этих комплексов. По А.Бонди |
||
|
молекулярный кристалл получается, когда закрыто |
||
|
более 80 % координационной сферы металла. Далее |
||
|
исследователи для количественной оценки степени |
||
|
пространственного экранирования центрального атома |
||
|
использовали телесные углы с вершиной на атоме |
||
|
металла, заполняемые заместителями. Атомы |
||
|
представляются |
в виде жестких шаров с |
|
соответствующими Ван-дер-Ваальсовыми радиусами. Общая идея расчета телесных углов заместителей и цилиндрических проекций молекул состоит в
построении проекции молекулы в сферической и
цилиндрической системах координат соответственно на сферу и цилиндр единичного радиуса.
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
12 |

_
Телесный угол, заполняемый заместителем при атоме металла (а), построение цилиндрической проекции молекулы (Ме3Gе)2Hg (б) и развертка цилиндрической проекции (ось цилиндра Ge–Ge) молекулы (Ме3Ge)2Hg (в).
•На рис.- след. слайд приведена общая схема определения телесных углов лигандов. Сумма телесных углов лигандов в молекуле характеризует степень заполнения координационного пространства
у атома М и тем самым возможность реализации в
конденсированном состоянии межмолекулярных взаимодействий лиганд-лиганд (R...R'), металл-
лиганд (М…R') или металл-металл (М…М' ).
Величина телесного угла, свободной от координации области вокруг
•
атома М, =4П-Q, где 4П=12,56 рад - максимальное
значение . Чем больше Q (и соответственно меньше ) тем вероятнее, что структура конденсированного состояния образована лишь взаимодействиями R...R' , но не М - R' или М - R' .
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
13 |

_
Методы оценки заполнения координационной сферы, привлекают своей наглядностью и позволяют не
прибегая к методам молекулярной
механики рассчитывать в нулевом приближении, например, возможные варианты синтеза координационнонасыщенных соединений. Правда, "метод
телесных углов" недостаточно учитывает
степень заполнения координационной сферы экранирующими атомами лигандов отдельно.
•Метод «Расширяющейся координационной сферы»
Более перспективным подходом является
представление степени заполнения координационной сферы у ц.а. в виде отношения части площади сферы (с центром, совпадающим с ц.а.), пересекающей лиганд, к
общей площади данной сферы в зависимости
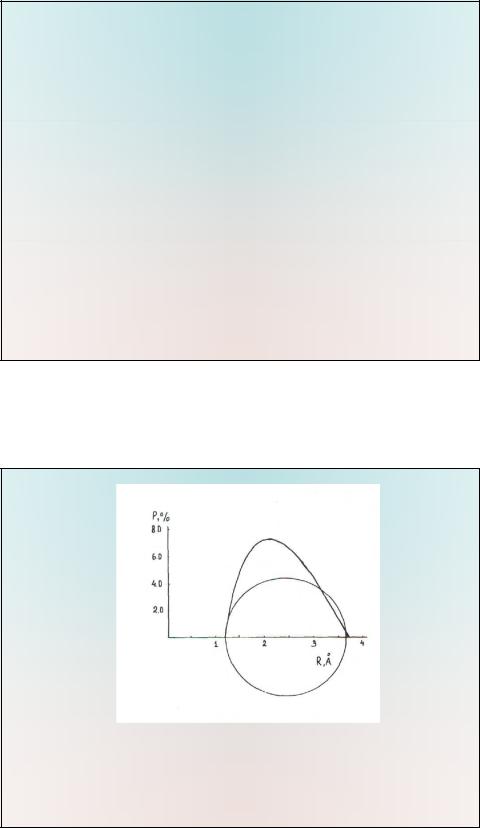
от ее радиуса R (рис. ниже). В этом случае по графику определяется "критическое сечение" лиганда Рmax. Сумма Pмах не может быть более 100%.
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
14 |
_
•Зависимость степени заполнения координационной сферы P(%) вокруг ц.а.(начало коорди-нат) от
расстояния до геометрического
центра лиганда (для случая сферического лиганда). Расстояние металл-лиганд RM–L=2,426 Ǻ, радиус Ван-дер-Ваальса RVDW=1,25 Ǻ,
Зависимость степени заполнения координационной сферы P(%) во-круг ц.а.(начало координат) от расстояния до геометрического центра лиганда (для случая сферического лиганда). Расстояние металл-лиганд RM–L=2,426 Ǻ, радиус Ван-дер-Ваальса RVDW=1,25 Ǻ,
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
15 |
_
•При этом наглядно видно, какие группы атомов не экранированы, видна и потенциальная возможность
модификации лиганда в рамках данного соединения, например,
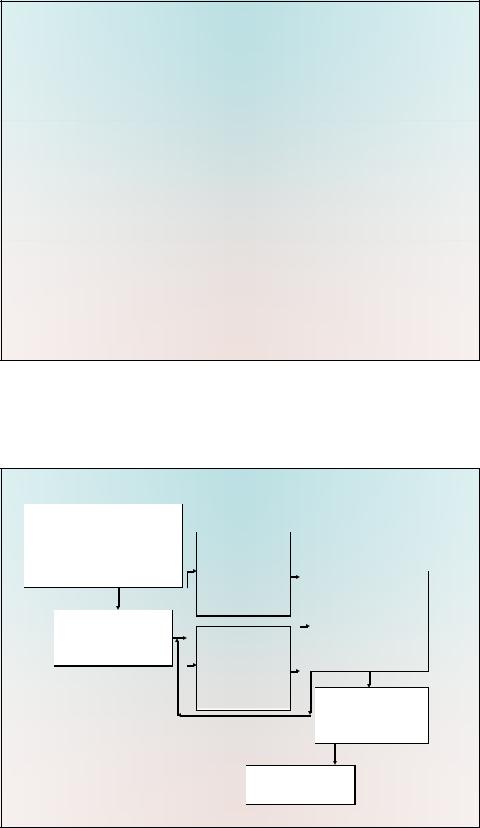
развитием его углеводородной периферии. Данный метод реализован в виде компьютерной программы «СФЕРА», блоксхема которой представлена на рис.
•В этом случае экранирование ц. а. и отдельных фрагментов гипотетической молекулы может быть определено следующим образом:
•1. С той или иной степенью приближения методом молекулярной механики конструируется изолированная
молекула соединения.
•2. Рассчитывается зависимость степени заполнения сферы от расстояния до геометрического центра лиганда.
•
Ввод данных: ортогональных координат атомов лигандов и их радиусов Ван-дер-Ваальса. Ц.а. расположен в начале координат.
Построение двух идентичных молекул соединения
Три |
вращения |
1 |
|
|
|
|
|
|
молекулы |
|
на |
|
|
|
|
|
|
произвольный угол |
|
|
|
|
|
|||
по |
трем |
осям |
|
|
|
|
|
|
|
|
Определение |
||||||
координат |
|
|
|
|
||||
|
|
|
|
минимального |
||||
|
|
|
|
|
|
|||
|
|
|
|
|
|
расстояния |
между |
|
|
|
|
|
|
|
молекулами |
(точки |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
наиболее |
вероятного |
|
Три |
вращения |
2 |
|
|
||||
|
|
контакта) |
|
|||||
молекулы |
|
на |
|
|
|
|
|
|
произвольный угол |
|
|
|
|
|
|||
по |
трем |
осям |
|
|
|
|
|
|
|
|
|
|
|
||||
координат |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Обработка |
данных |
|
|
|
|
|
|
|
(расчет |
вероятности |
|
|
|
|
|
|
|
||
следующий цикл |
контакта) |
|
Вывод данных (монитор, принтер)
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
16 |

_
В методе «случайных столкновений», реализованном в |
|||||
компьютерной |
программе |
«КОНТАКТ», |
|
рассматриваются |
|
случайные столкновения идентичных молекул соединения. Каждая |
|||||
молекула, при фиксированном положении второй, три раза |
|||||
поворачивается на произвольные углы относительно трех осей |
|||||
координат, после чего рассчитывают минимальное расстояние |
|||||
между атомами противоположных молекул, которое заносится в |
|||||
память компьютера. После большого числа таких циклов вращения |
|||||
(как правило 500) рассчитывается вероятность осуществления |
|||||
контакта того или иного атома молекулы. Атомы, у которых |
|||||
значение данной вероятности отлично от «0», считаются |
|||||
участвующими в межмолекулярном взаимодействии. Далее |
|||||
осуществляется разбиение атомов лигандов на функциональные |
|||||
группы и расчет энтальпии парообразования методом аддитивных |
|||||
вкладов. В данном подходе используется классическое понимание |
|||||
межмолекулярного контакта, то есть атомы двух молекул |
|||||
считаются контактирующими, |
если |
расстояние между ними |
|||
|
|
- |
- |
. |
, |
очевидно, что при сближении двух идентичных молекул наиболее вероятным будет контакт между атомами с наименьшем расстоянием между ними.
•Метод полиэдров «Воронова-Дирихле» Метод «полиэдров Воронова-Дирихле» использует стандартную кристаллографическую информацию и позволяет находить межмолекулярные контакты, не привлекая дополнительных данных, в том числе значений радиусов Ван-дер-Ваальса.
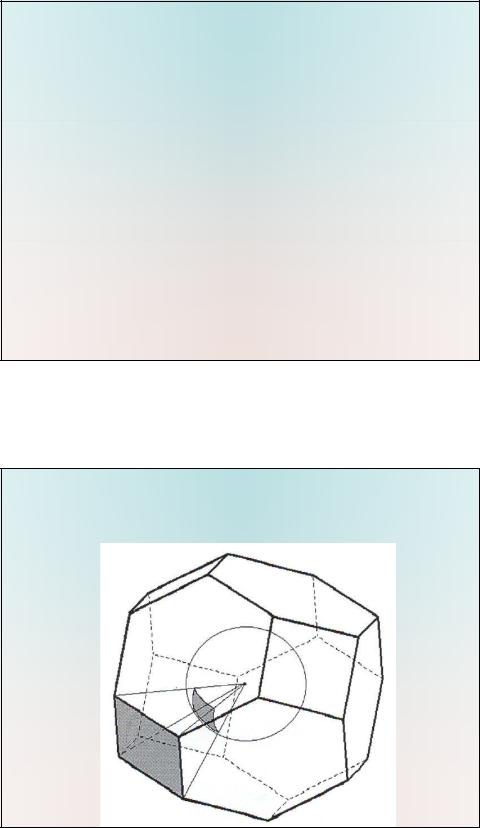
•Молекулярный полиэдр Воронова-Дирихле (ПВД) представляет собой область пространства, ограниченную набором пересекающихся плоскостей, каждая из которых перпендикулярна отрезку, соединяющему атом рассматриваемой молекулы с каким-либо атомом одной из молекул окружения, и делит этот отрезок в отношении, зависящем от природы контактирующих атомов. Для атомов одинакового сорта (например, атомы водорода) коэффициент деления равен 0.5. Каждая грань молекулярного ПВД отвечает контакту между атомами соседних молекул. Граничная поверхность, формируемая гранями ПВД атомов, отвечающих контактам между атомами двух соседних молекул, соответствует контакту молекула-молекула. Для оценки взаимодействия пары молекул относительная сила межмолекулярного взаимодействия оценивается суммой телесных углов Wi, соответствующих этим контактам, нормированной на сумму телесных углов для всех не валентных связей, образованных молекулой, принятой за центральную (WS), т.к. в общем случае между данной парой молекул существует несколько контактов: .
©ИОНХ РАН © Севастьянов В.Г.
Попов В.С. |
17 |
_
•Процедура построения молекулярного ПВД автоматизирована в рамках комплекса программ TOPOS и состоит из трех стадий:
•1. Построение ПВД для всех независимых атомов в элементарной ячейке;
•2. Классификация всех межатомных контактов, которым соответствуют грани ПВД. Выделение валентных и не валентных контактов;
•3. Идентификация молекулярных структурных единиц. Поиск внутри- и межмолекулярных контактов.
•Классификация межатомных взаимодействий осуществляется с использованием метода пересекающихся секторов. В рамках этого метода наличие контактов между атомами определяется в результате учета числа парных перекрываний сферы, описанной вокруг каждого атома рассматриваемой пары и имеющей радиус,
равный радиусу Слейтера (rs) соответствующего атома, с набором
сферических секторов, радиус (rsec) каждого из которых может быть найден из соотношения: Vp=1/3×W×rsec3, где Vp и W - соответственно объем, и телесный угол пирамиды, в основании которой лежит грань полиэдра ВД, отвечающая межатомному контакту, а в вершине - рассматриваемый атом (рис.2).
Полиэдр ВД атома. Телесный угол ( ) заштрихованной грани численно равен площади сегмента сферы единичного радиуса, высекаемого
пирамидой, опирающейся на эту грань.
© ИОНХ РАН © Севастьянов В.Г. |
|
Попов В.С. |
18 |

_
В рамках данного подхода внутренняя слейтеровская сфера отвечает максимуму электронной плотности в валентной оболочке атома, а фрагмент сферы, соответствующий сектору, ограничивает атом в конкретном кристаллическом поле в направлении межатомного контакта. В этом случае для любого межатомного контакта наблюдается, по крайней мере, одно перекрывание , которое отражает взаимодействие областей соседних атомов. Считается, что химическая связь возникает только между “прямыми” соседями при наличии, по крайней мере, еще одного перекрывания , указывающего на участие во взаимодействии валентных оболочек атомов .
•Роль центрального атома в процессах парообразования при выполнении условия достаточного экранирования
•Рассматривая роль центрального атома координационно-насыщенного соединения с молекулярным строением удобнее, хотя это и достаточно условно, рассмотреть две возможности влияния этого ключевого звена молекулы на энергетические характеристики ММВ – собственно межмолекулярное взаимодействие вида центральный атом
– молекула и влияние ц.а. на энергетические характеристики атомов, группы атомов данной молекулы, участвующих в ММВ.
•Впервые строгое и последовательное объяснение сил Ван-дер- Ваальса появилось в 1930 г. в работах Лондона, который свел их к электрическим взаимодействиям, Лондон ввел строгую формулу для энергии дисперсного взаимодействия пары молекул, исходя из квантово-механической теории возмущений второго порядка. Взаимодействующие молекулы рассматриваются как квазиупругие осцилляторы, энергия которых отлична от нуля. Энергия взаимодействия U, усредненная по различным ориентациям молекул относительно соединяющего их вектора R, изменяется обратно
пропорционально шестой степени расстояния между их центрами R:
(А - константа).
•Энергия U(R) - величина отрицательная, а следовательно, соответствующая сила Ван-дер-Ваальса - сила притяжения. Применив метод к группе молекул, Лондон получил сумму энергий взаимодействия частиц, взятых попарно. Иными словами, дисперсионный эффект обладает _свойствами аддитивности.
©ИОНХ РАН © Севастьянов В.Г.
Попов В.С. |
19 |

_
•Классификация атомов при расчетах энергетики молекул обычно проводится по атомам или по связям. В рассматриваемом варианте структурный элемент - центральный атом, для которого учитывается "первое окружение" - совокупность непосредственно связанных с ним атомов другого вида. Если взять связывающую часть составляющей энергии межмолекулярного взаимодействия U (по Лондону) можно показать, что для пары атомов уже при расстояниях между сферами Ван-дер-Ваальса ~0,2–0,З Ǻ составляющая энергии дисперсионного
взаимодействия будет менее 10-3 кДж/моль, т.е. практически
незначима.
•Это, в общем, проистекает и из понятия радиуса Ван-дер-Ваальса как радиуса сферы, ограничивающей область связывания данного атома с другим атомом.
•Таким образом, условия, при которых собственно ц.а. не участвует в Ван-дер-Ваальсовых взаимодействиях, могут определяться экранированием сферы ц.а. от межмолекулярных контактов. "Первое окружение" таким образом экранирует ц.а., нивелирует его физикохимическую индивидуальность с точки зрения ММВ. Однако
опосредованное влияние ц.а. может и должно проявляться на ММВ
через "первое окружение". Это влияние должно быть более ярко выражено в гомолептических ионных галогенидах, в меньшей степени в случае образования дополнительных связей атомами "первого окружения" ц.а.. Возможны случаи (ковалентносвязанные ароматические - системы), когда уже первое окружение (ураноцен - U(C8H8)2, ферроцен Fe(C6H6)2) нивелирует роль ц.а..
•С.С.Яровой отмечает, что геометрия структурного элемента связи с учетом первого окружения практически не зависит от того, какие атомы находятся во втором и последующих окружениях этой связи. В большинстве соединений любые два атома, расположенные далеко друг от друга в цепи молекулы, будут и пространственно далеки друг от друга. Этим в значительной степени объясняется то, что рассмотрение только
первого окружения каждой связи оказывается вполне
удовлетворительным при расчете физико-химических свойств данного типа соединений (алканов), причем в большинстве случаев точность расчета соответствует точности лучших экспериментальных данных.
•Ю. А.Лебедев применительно к задаче расчета физикохимических свойств органических соединений использовал схему по атомам с учетом первого окружения. Расчетные величины также прекрасно согласуются с экпериментальными. Таким образом, учитывая современный уровень экспериментальной техники определения энтальпий испарения и сублимации, можно полагать, что в случае достаточного экранирования ц.а., его влияние может, в принципе, проявляться в ММВ первого окружения ц.а. и несущественно при экранировании данного первого окружения от участия в ММВ.
©ИОНХ РАН © Севастьянов В.Г.
Попов В.С. |
20 |