

Лекция N 1
1.Предмет, структура и методы ф изической химии.
1.1.Физическая химия изучает общие вопросы химического сроения вещества,
общие количественные |
закономерности |
химического |
равновесия |
и |
химических процессов, |
а также ф изических явлений, |
которые |
их |
|
сопровождают. |
|
|
|
|
1.1.1. Термин ф изическая химия введен М.В .Ломоносовым. « Физическая химия - наука, которая должна на основе положений и опытов ф изических объяснить причину того , что происходит через химические операции в сложных телах.» Т. е. ФХ изучает связь между ф изическими и химическими явлениями, ф изические процессы, которыми сопровождаются химические реакции.
1.1.2..Физическая химия – это теоретическая химия. Которая рассматиривает общие законы, определяющие химические процессы, а именно направление хим. реакций, условия равновесия, протекание во времени, влияние различных ф акторов на химические реакции.
1.1.2.ФХ дает математическое описание химических процессов, что позволяет перевести изучение химических явлении на компьютерную основу.
1.1.3.Физическая химия – основа химической технологии, она позволяет оптимизировать протекание химических реакций, сделать их более экологически безопасными.
1.1.4.Физическая химия изучает строение веществ, связь между структурой и ф изико-химическими свойствами веществ, т.е. дает основу материаловедения, науки, которая определяет технический прогресс.
1.1.5.Физическая химия - основа аналитической химии, т.к. она имеет дело с
методами изучения структуры вещества, а, следовательно его идентиф икации, ф изическими методами.
1.2.Основные разделы ф изической химии
1.2.1.Химическая термодинамика – как часть научной дисциплины общей термодинамики – исследует условия равновесия в ф изико-химических процессах и химических реакциях.
1.2.2.С троение вещества –теории химической связи и расчетные и экспериментальные методы изучения строения.
1.2.3.Химическая кинетика – протекание во времени и механизмы химических реакций
1.2.4.Э лектрохимия Промежуточные, объединяющие разделы:
Статистическая термодинамика
Теория реакционной способности Термодинамика необратимых процесов
Электрохимическая кинетика
2.Основные понятия химической термодинамики
2.1.Термодинамические системы .
2.1.1.Термодинамическая система – это часть материального мира, отделенная от окружающей среды реальными или воображаемыми границами и являющаяся объектом исследования. Окружающая среда значительно больше по объему, и поэтому изменения в ней незначительны
http://www.mitht.org/forum/
по сравнению с изменением сосотояния системы. В отличие от механических систем, которые состоят их одного или нескольких тел, термодинамическая система содержит очень большое число частиц, что порождает совершенно новые свойства и требует иных подходов к описанию состояния и поведения таких систем. Термодинамическая система представляет собой м а к р о с к о п и ч е с к и й о б ъ е к т.
2.1.2. Классиф икация термодинамических систем 2.1.2.1. По составу
Термодинамическая система состоит из компонентов. К о м п о н е н т - это вещество, которое может быть выделено из системы и существовать вне ее, т.е. компоненты – это независимые вещества.
2.1.2.1.1.Однокомпонентные.
2.1.2.1.2.Двухкомпонентные, или бинарные.
2.1.2.1.3.Трехкомпонентные – тройные.
2.1.2.1.4.Многокомпонентные.
2.1.2.2.По ф азазовому составу – гомогенные и гетерогенные
2.1.2.2.1.Г о м о г е н н ы е системы имеют одинаковые макроскопические свойства в любой точке системы, прежде всего температуру, давление, концентрацию, а также показатель преломления, диэлектрическую проницаемость, кристаллическую структуру и др.. Гомогенные системы состоят из одной ф азы. Ф а з а – это однородная часть системы, отделенная от других ф аз поверхностью раздела и характеризуеемая своим уравнениеи состояния. Фаза и агрегатное состояние – не перекрывающиеся понятия. Агрегатных состояний только 4 , ф аз может быть гораздо больше.
.1.2.2.2. Г е т е р о г е н н ы е системы состоят минимум из двух ф аз.
2.1.2.3.По типам связей с окружающей средой (по возможностям обмена с окружающей средой
2.1.2.3.1.Изолированная система не обменивается с окружающей ни энергией , ни веществом.
2.1.2.3.2.Закрытая система может обмениваться с окружающей средой энергией, но не обменивается веществом.
2.1.2.3.3.Открытая система обменивается и энергией, и веществом
2.2.С остояние ТДС
2.2.1.Определение. С остояние ТДС – это совокупность всех ее макроскопических свойств, обычно имеющих количественное выражение. Макроскопический характер свойств означает, что их можно приписать только к системе в целом, а
не отдельным частицам, которые составляют ТДС ( Т, р, V, c, U, ni). Количественные характеристики состояния связаны между собой. Поэтому существует м и н и м а л ь н ы й набор характеристик системы, называемых п а р а м е т р а м и, задание которых позволяет полностью описать свойства системы. Количество этих параметров зависит от типа системы. В простейшем случае для закрытой гомогенной газовой системы в состоянии равновесия достаточно задать только 2 параметра.
2.2.2.Р авновесие
Классическая термодинамика обычно ограничивается рассмотреним равновесных состояний ТДС . Р а в н о в е с и е - это такое состояние, к которому самопроизвольно приходит ТДС , и в котором она может существовать бесконечно долго в отсутствии внешних воздействий. Для определения
http://www.mitht.org/forum/
равновесного состояния всегда требуется меньшее количество параметров, чем
для неравновесных систем.
2.2.3. У р а в н е н и е с о с т о я н и я связывает параметры состояния и относится к фазе. Общий вид его f (p,V, T) = 0. Простейший вид уравнения состояния имеет идеальный газ
pV = nR T, где R = 8.314 Дж/(моль.К) = 1,987 кал/(моль.К) = 0,082 л.атм/(моль.К) .
[p] = Па, 1атм = 1,013*105 Па, [V ] = м3, 1 м3 = 103 л,
[T]= К,
[n]= моль, N = 6.02*1023 моль-1= 6,02*1026 кмоль-1 .
2.2.4.Функции состояния – это характеристики системы, которые зависят только от параметров состояния, но не зависят от способа его достижения.
|
F (x,y) |
F(V,T] |
|
|
|
p,V,T |
∫dF = 0 |
dF – полный диф ф еренциал |
|||
|
dF = ( |
∂F |
)T dV + ( |
∂F |
)V dT, |
|
∂V |
dT |
|||
|
|
|
|
||
2
∫dF = F 2 – F 1 = ∆F – изменение ф ункции состояния не
1
зависит от пути перехода, а определяется только начальным и конечныи состоянием, ∆F – всегда разность между конечным и начальным сосотянием. 2.2.5. Классиф икация параметров и ф ункций состояния 2.2.5.1. Э кстенсивные свойства при объединении (контакте) двух систем складываются
X = X’ + X’’
( Объем, число молей, энергия. теплоемкость )
Э кстенсивные свойства зависят от числа молей веществ. Чтобы описать экстенсивные свойства, их относят к определенному количеству вещества, например к одному молю
X/n = X - мольные величины,
к единице массы X/m = X уд. – удельные величины, к единице объема X/V = ρ - плотности.
б) Интенсивные свойства при контакте двух систем выравниваются ( температура, давление, поверхностное натяжение, электрический потенциал)
Y ’1 |
< Y’’1 |
Y 2 = Y’2 = Y’’2 , |
Y ’1 < Y 2 < Y’’1. |
2.3. Термодинамический процесс изменение состояния системы при взаимодействии с окружающей средой
2.3.1. Классификация термодинамических процессов
2.3.1.1.Циклические и нециклические
2.3.1.2.Обратимые и необратимые
http://www.mitht.org/forum/

2.3.1.2.1.О б р а т и м ы е процессы проходят в прямом и обратном направлении через одни и те же промежуточные состояния, и после завершения цикла ни в системе, ни в окружающей среде не наблюдатся никаких изменений. Обратимые процессы называют часто р а в н о в е с н ы м и, так как в каждый момент промежуточного состояния внешние и внутренние силы должны быть скомпенсированы (это характерно для состояния равновесия), точнее они должны отличаться на бесконечно малую величину. Такой процесс происходит бесконечно медленно, т.е. время не учитывается в термодинамике обратимых процессов, и их также называют к в а з и с т а т и ч е-с к и м и. Пример – обратимое изотермическое расширение идеального газа. В обратимом процессе работа полученная в прямом направлении должна быть численно равна работе, затраченнойв обратном процессе.
2.3.1.2.2.Необратимые процессы в прямом и обратном направлении идут через разные промежуточные состояния. Р абота, получаемая в необратимом процессе всегда меньше работы обратимого процесса при заданных начальном и конечном состоянии.
2.3.2.Теплота и работа - способы обмена энергией между ТДС и окружающей средой
Теплота и работа характеристики процесса, они не являются ф ункциями состояния
2.3.2.1.Теплота – ф орма обмена энергией на микроскопическом уровне, т.е. в
форме изменения хаотического движения молекул. Теплота считается положительной, если она подводится к системе.
2.3.2.2. Р абота - ф орма обиена энергией на макроскопическом уровне, когда происходят направленное перемещение объекта. Р абота считается положительной, если ее совершает система против внешних сил.
http://www.mitht.org/forum/

Лекция N 2
Взависимости от характера объекта различают разные виды работы:
2.3.2.2.1.Механическая - перемещение тела
δW мех = - Fdl,
2.3.2.2.2. Расширение газа
δW = рdV pe = pi в обратимом процессе. 2.3.2.2.3. Э лектрическая – перемещение электрических зарядов
δW эл = -ϕdq
2.3.2.2.4. Поверхностная – изменение поверхности
δW поверхн. = -σdS
2.3.2.2.5. Общее выражение для работы δW = - Ydx
Y – обобщенная сила на обобщенную координату или произведение интенсивного ф актора на изменение экстенсивного.
2.3.2.2.6. Все виды работы кроме работы расширения называются п о л е з н о й работой(δW’).
δW = рdV + δW’
2.3.2.2.7. По аналогии можно ввести понятие х и м и ч е с к о й работы, когда направленно перемещается химическое вещество – экстенсивное свойство, при этом интенсивный параметр называется химическим потенциалом
δW хим = -Σµkdnk.
2.3.3.Нулевой закон термодинамики. С ф ормулирован позже первого и второго, но по логике построения постулатов термодинамики он должен предшествовать им. Е сли две системы находятся в тепловом равновесии с третьей, то они находятся и в тепловом равновесии меджу собой.
Т А = Т В ; Т А = Т С Т В = Т С .
Таким образом ответственным за тепловое равновесие является интенсивный параметр - температура. Поэтому можно процессы теплообмена также представить в виде аналогичной по структуре ф ормулой
Q = TdS ,
где экстенсивное свойство называется энтропией, а знак + соответствует передаче энергии системе в тепловых процессах
3.Первый закон термодинамики
3.1.Содержание первого закона термодинамики
3.1.1. Первый закон термодинамики является частным случаем закона
сохранения энергии. Эмпирический закон Майер (1842), Джоуль (1842), Гесс (1840), Гельмгольц (1847). Энергия не создается и не уничтожается, а только переходит из одной формы в другую. Невозможен вечный двигатель
1-го рода, который работал бы без подвода энергии.
3.1.2. Внутренняя энергия как функция состояния системы
Циклический процесс |
|
Нециклический процесс |
p1 V1, T1 |
|
p1 V1, T1 |
Q = W |
Q1 ≠ W1 |
Q2 ≠W2 |
http://www.mitht.org/forum/

P 2 V 2 , T 2 |
p2 V 2 , T 2 |
Q 1 + Q 2 = W 1 + W 2
Q 1 – W 1 == -(Q 2 – W 2 ) = = -(Q 3 – W 3 )
Р азность мекжду теплотой и работой при заданных начальных и конечных параметрах системы является величиной постоянной и не зависит от пути процесса, т.е. представляет собой ф ункцию состояния системы. Клаузиус назвал ее изменением внутренней энергией.
∆U = Q – W = U 2 – U 1 . ∫dU = 0 .
Или в диф ф еренциальной ф орме dU = δQ - δW
Внутренняя энергия – это сумма различных видов энергии, т.е. кинетическую и потенциальную энергию частиц, составляющих систему, но не включает кинетической энергии системы.
3.1.3. Математическое выражение и ф ормулировка 1-го закона Q = ∆U + W
Тепло, подведенное к системе, идет на изменение внутренней энергии системы и совершение ею работы против внешних сил
δQ = dU + δW = dU + pdV + δW’
Классическая термодинамика не дает способов расчета и измерения внутренней энергии, а только ее измннение.
3.2. Применение 1-го закона к простейшим системам 3.2.1. Изолированная система
Q = 0 , W = 0 ., ∆U = 0. Внутренняя энергия изолированной систкемы постоянна.
3.2.2.Изохорный процесс V = const.
3.2.2.1.Теплота изохорного процесса
pdV = δW расширения = 0. W расширения = 0.
Е сли δW’ = 0, δQ = dU Q v = ∆U
В отсутствие полезной работы изменение внутренней энергии равно теплоте изохорного процесса.
3.2.2.2. Изохорное нагревание |
∂U |
|
||||
δQ = n C vdT = dU : n dU = |
C |
vdT C v |
|
= ( |
)v |
|
|
∂T |
|||||
m cуд dT/ |
|
|||||
|
|
|||||
3.2.3. Изобарный процесс p = const |
|
|
||||
3.2.3.1. Р абота изобарного процесса |
|
|
||||
2
δW = ∫ pdV = p (V 2 – V 1 ) = p ∆ V
1
3.2.3.1. Теплота изобарного процесса
Q p = ∆U + p ∆ V = ( U 2 + pV 2 ) – (U 1 – pV 1 ) = H 2 – H 1 = ∆ H
http://www.mitht.org/forum/
Q p = ∆H
3.2.3.3. Э нтальпия – ф ункция состояния
H ≡ U + pV
В энтальпию входит внутренняя энеогия и произведение параметров Полный диф ф еренциал dH = dU + pdV + Vdp.
При р= const dH = dU + pdV = Q p. 3.2.3.4. Изобарическое нагревание
δQ p = n C p |
|
|
|
dT = dH |
|
|
|
∂H |
)p |
|
dT = dH, C p |
|
C p |
|
= |
|
|||
|
|
∂T |
|||||||
|
|
|
|
|
|
|
|
|
3.2.4.Изотермический процесс Т =const 3.2.4.1.Изотермическое расширение идеального газа
|
U = f(T). |
dU =0 |
|
|
|
|
|
δQ = δW расшир. = pdV; |
|
|
|||
2 |
|
|
V 2 |
|
||
Q = W = ∫pdV = ∫ nR T dV |
= nR T ln |
= nR T ln |
||||
V |
|
|||||
1 |
V |
|
1 |
|
||
|
|
|
|
|||
p1
p2
3.2.5. Адиабатический процесс
Q = 0; δQ = 0; 0 = dU + δW расшир; δW расшир = - dU; W = - ∆U
Р абота совершается за счет убыли внутренней энергии, например за счет охлаждения системы
2
dU = n C vdT; W = - ∫nC V dT = -nC v ∆T, если C v = сonst.
1
Е сли над системой соверщается работа (сжатие, пропускание электрического тока, введение новых веществ, то внутренняя энергия системы увеличивается.
4. Термохимия
4.1. Предмет Термохимия занимается тепловыми эф ф ектами, сопровождающими
химические и ф изико-химические процессы, т.е измерениями теплоты, которая выделяется или поглощается в химический реакциях, ф азовых переходах или образовании и разбавлении растворов.
4.2.Понятие теплоты химической реакции
4.2.1.С техиометрическое уравнение химической реакции и химическая переменная.
Обычнаяч запись уравнения химической реакции в общем виде
νA A = νB B = νC C + νD D, или в общем виде, 0 = ∑νk A k ,
k
где νk– с т е х и о м е т р и ч е с к и й к о э ф ф и ц и е н т с у ч е т о м з н а –к а : для реагентов он отрицателен, а для продуктов положителен. Э то
очень удобно для обработки на компьютере.
C техиометрическое уравнение связывает изменения чисел молей всех веществ, участвующих в реакции
3H 2 + N2 = 2NH 3
http://www.mitht.org/forum/
νi -3 |
-1 |
2 |
|
|
|
|
dn H 2 |
= − |
dn N 2 |
= |
dn NH 3 |
= |
dn i = dξ. |
3 |
1 |
|
2 |
|
νi |
|
ξ- называется химической переменной, степенью превращения, глубиной протекания реакции, координатой реакции, и является важнейшей характеристикой протекания реакции в закрытой системе, так как все изменения количества различных веществ могут быть описаны изменением одного параметра.
С помощью нее можно составить уравнения материального баланса в закрытой системе с химической реакцией.
g |
ξ |
|
Уравненние материального баланса в |
∫dn k |
= ∫νk dξ |
nk = nk0 + νk ξ |
закрытой системе. |
n 0 |
0 |
|
|
|
|
|
|
http://www.mitht.org/forum/

1
Лекция № 3
4.2.2. Теплоты химических реакций и их связь с изменением термодинамических
функций.
4.2.2.1.Теплота при постоянном оъеме
Q v = ∆U,
Е сли W’ = 0, то U = f (T, V, ni)
Q v = ∆rU, при V=const, T = const, W’ = 0, ξ = 1.
Теплотой химической реакции называют количество тепла которое выделяется или поглощается при полном превращении реагентов в продукты в мольных количествах, соответствующих стехиометрическим коэф ф ициентам, при постоянном объеме, постоянной температуре и отсутствии полезной работы.
4.2.2.2. Теплота при постоянном давлении
Q р = ∆r Н, при р=const, T = const, W’ = 0, ξ = 1.
4.2.3. Классиф икация реакции по знаку теплового эф ф екта
∆U = Q v > 0
∆Н = Q р > 0 реакция эндотермическая
∆U = Q v < 0
∆Н = Q р < 0 реакция экзотермическая 4.2.4. С вязь Q р и Q v в химических реакциях ∆Н = ∆U + p ∆ V
В химических реакциях объем изменяется, если изменяется число молей газообразных веществ
p∆ V = ∆nгаз R T.
∆nгаз = ( ∑νj − ∑ |
|
νi |
|
)газ = ∆ ν k газ = ∑νk газ |
|
|
|
|
|
|
|
||||
j |
i |
|
k |
|
|
||
Пример |
|
|
|
|
|
|
|
|
|
|
|
|
1/2N2 + 3/2 H 2 = NH 3 |
|
|
|
|
|
|
|
νk -1/2 -3/2 |
1 |
∆ν = -1 |
∆H = -46, 19 кДж/моль = Q p
Q V = Q p + R T = -46190 + 8,314 298 = -43710 Дж/моль = - 43, 71 кДж.моль
4.3.Закон Гесса (1836)
4.3.1.Формулировка Теплота реакции не зависит от пути протекания процесса, т.е. промежуточных
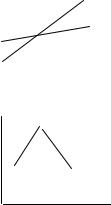
химических стадий, через которые он проходит, а только от начального и конечного состояния веществ. Закон Гесса вкладывает дополнительный смысл в стехиометрическое уравнение реакции, а именно, что каджому веществу соответствует определенный уровень энергии и разность между этими уровнями определяет количество тепла, выделяемое или поглощаемое в химических реакциях, можно сказать, что вещества образуют потенциальное поле, в котором переходы не зависят от пути процесса.
4.3.2.Изменение внутренней энергии и энтальпии системы в химической реакции
Q V = ∆rU = ∑νj U j − ∑ νi U i = ∑νk U k .
j |
|
k |
Q p = ∆rH = ∑νj H j − ∑ νi H i = ∑νk H k ,
j |
|
k |
где индекс i относится к реагентам, индекс j – к продуктам, а индекс k –ко всем веществам, участвующим в реакции.
http://www.mitht.org/forum/

2
4.3.3. Применение закона Гесса. Закон Гесса позволяет комбинировать стехиомет рические уравнения, вкладывая в них термохимический смысл. Любые линейные комбинации с одинаковыми общими уравнениями имеют один и тот же тепловой эф ф ект. Э то позволяет легко определять теплоты реакций, которые трудно экспериментально провести до конца в соответствие с требованиями
|
|
|
|
|
|
|
|
H 2 + O 2 = H 2 O 2 |
+1 H 2 + 1/2 O 2 = H2 O |
∆rH 1 = -285.8 кДж/моль |
|||||||
-1 H 2 O 2 |
= H |
2 O + 1/2O 2 |
|
∆rH 2 = -98,7 кДж/моль |
||||
|
|
|
|
|
|
|
||
|
|
H 2 + O 2 = H 2 O 2 |
∆ |
rH = ∆rH 1 - ∆rH 2 = -285,8 – (-98,7) = -187,1 кДж/моль |
||||
Э нергетическая диаграмма |
||||||||
_________________H 2 + O 2 |
||||||||
|
|
- |
187.1 |
|
|
С оставление циклов |
||
|
|
|
|
|
||||
|
|
-285.8 _______H 2 O 2 |
|
|
||||
|
|
- |
98.7 |
|
|
|
||
|
|
_________________H 2 O + 1/2 O 2 |
||||||
|
||||||||
4.4.Первое следствие из закона Гесса
4.4.1. Понятие стандартной теплоты образования вещества Так как невозможно измерить абсосютное значение энергии, а только ее изменение,
нужно выбрать для различных веществ условный 0, относительно которого определяются изменения внутренней энергии. За такой уровень отсчета приняты состояния простых веществ при стандартных условиях. Простые вещества образованы из атомов одного элемента. Е сли элемент существует в нескольких простых ф ормах, то выбирается наиболее устойчивая модиф икация
H 2 O 2 , N2 , Ar, C граф , Р красный.
С т а н д а р т н а я т е п л о т а о б р а з о в а н и я -это тепловой эф ф ект реакции образования вещества из простых веществ в стандартных состояниях. Обозначаеся ∆fH0 или ∆fU0, для простых веществ ∆fH0=∆fU0= 0.
Пример
C граф , + 1/2 O 2 + 3 H2 = C 2 H 5 OH (жидк.)
4.4.2.С тандартное состояние –наиболее устойчивая ф орма чистого вещества, для газов – давление 1 атмосф ере и газ должен обладать свойствами идеального газа, т.е. для него справедливо уравнение Менделеева-Клапейрона, конденсированные ф а- зы находятся в равновесии со своим паром. Температура не входит в понятие стандартного сосотояния. Оно возможно при любой температуре. В справочниках теплоты оюразования приводятся при температуре 298 К, но это специально оговаривается
∆fH0 298 .
4.4.3.1-го следствие из закона Гесса используется для определения теплот хи-
мических реакций по справочным данным, т.е. по теплотам образования ве-
ществ |
0 = ∑νk A k |
= 0 ; ∑ |
|
νi |
|
A i |
= ∑νj B j ∆rH0-? |
||||||||||
|
|
||||||||||||||||
|
|
|
|
|
|
k |
i |
|
|
|
|
j |
|||||
|
|
|
|
|
|
|
|
∆fH0 |
|
|
|
|
|||||
|
Р еагенты |
|
|
|
|
|
|
|
|
|
Продукты |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
Простые вещества |
|
|
|
|
|
|||||
|
− ∑ |
|
νi |
|
∆f H 0 i |
|
|
|
|
|
|
∑νj ∆f H j 0 |
|||||
|
|
|
|
|
|
|
|
|
|||||||||
|
i |
|
|
|
|
|
|
|
|
|
|
|
|
j |
|
|
|
http://www.mitht.org/forum/
3
∆rH0 = ∑νj ∆f H j 0 − ∑ |
|
νi |
|
∆f H 0 i = ∑νk ∆f H k 0 |
||
|
|
|||||
j |
|
i |
|
|
|
k |
Теплота реакции равна сумме теплот образования конечных веществ минус сумма теплот образования исходных веществ с учетом стехиометрических коэф ф ициентов по модулю.
4.5. Э кспериментальное определение теплот реакций. Теплоты сгорания. 4.5.1. Калориметрия – метод непосредственного экспериментального измерения
теплот химических реакций в приборе, называемом калориметром. Р азличают калориметры изотермические, работающие при постоянной температуре, и с переменной температурой, которая и служит измеряемой величиной в этом случае.
Р еакцию проводят в адиабатических ( по возможности) условиях. В этом случае изменение ∆U или ∆H происходят за счет химической реакции и за счет изменения темпе-
ратуры 0 = ∆rH + ∆tH = ∆rH ∆ξ + C p∆T ∆rH = - C p∆T/∆ξ = - C p∆T/ ∆n;
4.5.2. Теплота сгорания.
Для экспериментального определения теплоты реакции необходимо, чтобы реакция протекала однозначно, т.е. не сопровождалась побочными процессами и степень превращения приближалась к единице. Далеко не все реакции отвечают таким критериям. Наиболее доступны для экспериментального исследования теплоты сгорания, которые проводят в калориметрической бомбе в атмосф ере кислорода без катализатора. При этом получаются устойчивые оксиды С О 2, Н 2 О, S O 2, N2 .
С 2 Н 5 ОН +7/2 О 2 = 2 С О 2 + 3 Н 2О ∆с Н = Требуется анализ продуктов.
4.5.3.2-ое следствие из закона Гесса –определение теплоты реакции по теплотам сгорания
С 6Н 6 + 3Н 2 = С 6 Н 12 |
|
∆с Н 1 = -3267,58 кДж/моль |
|||||||
∆с Н 1 |
|
∆с Н 2 |
∆с Н 3 |
|
∆с Н 2 |
= - ∆fН воаы = -285,83 кДж/моль |
|||
|
|
|
|
|
|
|
|||
|
|
|
6 С О 2 + 6 Н 2 О |
|
|||||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
∆с Н 3 |
= -3919,91 кДж/моль |
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
||||
∆rН = ∑ |
|
νi |
|
∆сH 0 i - ∑νj ∆сH j 0 = - ∑νk ∆сH k 0 |
|
||||
|
|
||||||||
i |
|
|
|
j |
|
|
k |
|
|
|
|
|
|
|
|
|
|
|
|
Теплота реакция равна сумме теплот сгорания реагентов минус сумма теплот сгорания продуктов с учетом стехиометрических коэф ф ициентов.
4.5.4.3-ье следствие из закона Гесса – пересчет экспериментальных теплот сгорания на теплоты образования
|
|
3 С граф . + 3 Н 2 + 1/2О 2 = Н 3 С -С О-С Н 3 |
∆fH -? |
|
-1 |
|
C 3 H 6 O + 4 O 2 |
= 3 C O 2 + 3 H 2 O |
∆с Н = -1785.3 кДж/моль |
|
||||
3 |
|
Н 2 + 1/2О 2 |
= Н 2 О |
∆с Н = -285,8 кДж/моль |
3 |
|
С + О 2 |
= С О 2 |
∆с Н = -393,51 кДж/моль |
∆fH = сумме теплот сгорания простых веществ – теплота сгорания самого вещества
∆fH = ∑ νi ∆сH 0 i - ∆с Н
i
4.6Приближенный расчет теплот химических реакций по знергиям связи
4.6.1.Э нергия связи – это средняя энергия, необходимая для разрыва связи в 1 моле газообразного вещества до состояния газообразных атомов
|
|
HC lгаз = H + С l |
|
|
H 2 O газ = 2 Н + О |
H 2 O газ = Н + ОН |
∆H = 497 кДж/моль |
|
ОН = О + Н |
|
∆H = 421 кДж/моль |
|
http://www.mitht.org/forum/ |
|

4
εОН = 918/2 = 459 кДж/моль εОН 467 кДж/моль Э нергия ковалентных химических связей в сложных молекулах в первом приближении
можно считать величиной аддитивной, т.е. энергии связи независимы.
|
|
|
|
|
4.6.2. Р асчет теплоты образования вещнства по энергиям связи |
|||||||
3 |
С граф . + 3 Н 2 + 1/2О 2 = Н 3 С -С О-С Н 3 ж |
|||||||||||
∑ |
|
νi |
|
∆H атом. |
|
|
|
|
- ∆Hисп |
|||
|
|
|
|
|
|
|||||||
|
||||||||||||
|
i |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
С газ. + 6 Н + О = Н3С -С О-С Н3 газ |
|||||||||||
∆fH0 = ∑ |
|
νi |
|
∆ H атомизации - Σ npεp - ∆Hисп. |
||||||||
|
|
|||||||||||
|
|
|
|
|
i |
|
|
|
|
|
|
|
4.7.Теплота растворения Р аствор – это ф аза переменного состава, существующая в определенных пре-
делах термодинамических параметров. Р аствор – гомогенная система, состоящая из двух или более компонентов. С войства раствора зависят от концентрации, и теплота образования раствора также является ф унуцией концентрации. В процесс растворения вещества вносят вклад процессы разрушения кристаллической решетки (Q>0) и процесс сольватации (Q<0)
4.7.1.Интегральная теплота растворения – количество теплоты, выделяемое или поглощаемое при растворении 1 моля вещества в таком количестве водыБ чтобы была достигнута заданная концентрация раствора (моляльной) ∆Hm
∆H0
∆Hm
∆Hs
m, моль/1000г растворителя
4.7.2.Диф ф еренциальная теплота растворения вещества – это количество тепла выделяемое или поглощаемое при растворении 1 моля вещества в таком большом количестве раствора заданной концентрации, чтобы при этом концентрация раствора не изменялась.
4.7.3.Теплота разбавления раствора от одной концентрации до другой равна разности
между интегральными теплотами растворения
∆Hm12 = ∆Hm2 - ∆Hm1
4.7.4.Теплоты образования ионов в растворе
1/2Н2 + 1/2C l2 = HC lгаз
HC l + aq = HC laq = Н+aq + C l-aq
|
|
|
|
1/2Н2 + 1/2C l2 + aq= Н+aq + C l-aq |
∆H = ∆H1 + ∆H1 |
||
1/2Н2 + aq = Н+aq |
∆H1 = 0 |
|
1/2C l2+ aq = C l-aq ∆H2= ∆H |
http://www.mitht.org/forum/
1
Лекция № 4
4.7.Зависимость теплоты реакции от температуры
Р еагенты |
Т 1 |
Продукты Т 1 |
∆H т1 |
||
Р еагенты |
|
|
Т 2 |
Продукты Т 2 |
∆H т2 |
|
|||||
4.8.1.Зависимость теплоемкости от температуры для идеального газа
4.8.1.1. С вязь С р |
и С v |
|
|
|
|
∂U |
|
∂H |
|
С v = |
|
С p = |
∂T |
|
|
∂T v |
|
p |
|
С вязь С р |
|
и С v |
легко определяется для идеального газа: |
||||||
|
|
|
|
dH= dU + p dV = dU + p R /p *dT = dU + R dT |
|||||
|
|
|
|
|
|
(V |
= R T/p dV = R /p dT) |
||
|
∂H |
= |
|
∂U |
+ R |
|
C p |
|
= C V + R |
|
|
|
|
|
|||||
|
∂T p |
|
|
∂T v |
|
|
|
|
|
R – работа расширения, которую совершает 1 моль идеального газа при нагревании на 1о .
4.8.1.2. Э нергия молекул как сумма энергий различного вида движения Внутренняя энергия – это сумма энергии частиц, составляющих термодинамическую систему. В идеальном газе эта отсутствует энергия взаимодействия молекул, поэтому внутренняя энергия определяется только кинетической составляющей, но молекулы совершают различные виды движения, и приближенно общую кинетическую энергию
молекул можно представить в виде суммы энергии этих видов движения ε = εэлектр, +
εкол, + εвр. + εпост. = εе + εv + εr + εt.
Известно, что энергия микрочастиц квантована, т.е. может принимать только дискретные значения. Р асстояние между этими уровнями различно для разных типов движения ∆εе >>∆εv>>∆εr > ∆εt
Поступательная энергия молекул |
|
|
|
|
|
практически не квантуется, т.е. |
∆εе |
|
|||
может изменяться непрерывно |
|
||||
Когда системе сообщается тепло, |
|
||||
то энергия передается через хао- |
|
|
|
|
|
|
|
||||
тические соударения молекул. |
∆εv |
|
|||
При столкновениях молекулы |
|
|
∆εr |
|
kT |
|
|
|
|||
обмениваются энергией, величина которой зависит от температуры – « тепловыми квантами» - kT, где k – посотянная Больцмана = R /N = 1,38*10-16 Дж/К.
При обычной температуре величина теплового кванта достаточна, чтобы изменить энергию поступательного и вращательного движения, а также наиболее слабых колебаний, но возбуждения сильных колебений, и тем более электронов не происходит
∆εv>> kT > ∆εr.
Эта диаграмма позволяет примерно оценить теплоемкость простейших веществ
4.8.1.3.Одноатомный газ (He, Ar)
Молекулы одноатомного газа как точечные массы совершают только поступательное движение и имеют три степени свободы. По принципу равномерного распределения энергии по степеням свободы можно определить энергию молекул. На одну степень
свободы приходится в среднем ε = 1/2kT, а для 1 моля Е = 1/2R T. C ледовательно, на 3 степени свободы одноатомной молекулы приходится
|
|
U = 3/2 R T. |
|
∂U |
= 3/2 R ~ 3 кал/моль*K = 12,5 дЖ/мольК, C p = C V + R = 5 кал/мольК |
С v = |
|
|
|
∂T v |
http://www.mitht.org/forum/ |

2
Теплоемкость практически не зависит от температуры (кал/моль К)
Вещество |
nt |
nr |
nv |
150 K |
300 К |
400 К |
Ar |
3 |
0 |
0 |
2,92 |
2,98 |
2,98 |
O 2 |
3 |
2 |
1 |
4,98 |
5,03 |
6,22 |
H 2 O |
3 |
3 |
3 |
- |
6,04 |
7,57 |
4.8.1.4. Двухатомный газ – линейная молекула
Каждая молекула имеет 3N степеней свободы, где N –число атомов в молекуле. Для двухатомной молекулы общее число степеней свободы равно 6, из них 3 поступательных, 2 вращательных и 1 колебательная. Вращательных степеней свободы у линейных молекул только 2, так как привращении вокруг линии связия,
молекула не изменяет своего положения и это движение не может изменяться за счет передачи энергии от другой молекулы. При комнатной температуре возбуждаются только поступательное и вращательное движение – 5 степеней свободы
U = 5/2 R T.
|
∂U |
= 5/2 R ~ 5 кал/моль*K = 20,8 дЖ/мольК, C p = C V + R = 7 кал/мольК, |
|||
С v = |
∂T |
|
|||
|
v |
|
|
|
|
При повышении температуры начинает постепенно возбуждаться колебательное движение С v = 6 кал/моль К.
4.8.1.5. Многоатомные молекулы. Общее число степеней свободы равно 3N, из них 3 поступательных, 3 ( или 2 для линейных молекул) врашательных и 3N - 6 (5) колебательных. Колебания тоже могут быть разными валентные (жесткие) колебания, в которых происходит изменение длины связи и требующие большой энергии для возбуждения. Другие колебания деф ормационные, в них изменяются углы между связями. Они более мягкие и требуют меньших квантов для возбуждения, и следовательно могут возбуждаться при более низкой температуре.
Вобщем можно сделать такой вывод: чем сложнее молекула, тем сильнее зависимость ее теплоемкости от температуры. Нельзя получить общей теоретической ф ормулы, выражающей эту зависимость.
4.8.2. Теплоемкость твердых тел
Втвердом теле единственным видом движения является колебания элементов кристаллической решетки, ячейки которой повторяются в пространстве. Твердое тело можно представить как набор гармонических осцилляторов, Исследуя такую модель, Э йнштейн вывел теоретическую ф ормулу для расчета теплоемкости твердого тела на
основании статистической термодинамики (θ/T)2 e θ/T
С v |
|
= 3R |
|
|
θ = hν/k |
|
|
||||
|
|
(e θ/T –1)2 |
|
||
При низкой температуре θ/T -> , a теплоемкость -> 0 |
|||||
При высокой температуре |
θ/T -> 0, a теплоемкость -> 3R |
||||
Закон Дюлонга и Пти С v = суд. А = 6,3 кал
При очень низких температурах вблизи абс. нуля справедлив закон Дебая С v = а Т3
4.8.3.Представление температурной зависимости теплоемкости в виде температурного ряда. Так как теоретических общих уравнений для зависимости теплоемкости от температуры нельзя вывести, то используются
экспериментальные зависимости в виде степенного ряда Для органических веществ С р = а + bТ + сТ2 + dT3;
Для неорганических веществ С р = а + bТ + с'Т-2.
4.8.4.Уравнение Кирхгоф ф а – зависимость теплоты реакции от температуры
Q V = ∆rU = ∑νj U j − ∑ νi U i = ∑νk U k .
j |
|
k |
Q p = ∆rH = ∑νj H j − ∑ νi H i = ∑νk H k ,
j |
|
k |
http://www.mitht.org/forum/
3
∂∆ U |
|
|
|
∂U |
|
|
|
∂U |
|
|
∂U k |
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
− ∑[νi |
] |
|
|
|
|
|
|
|
|
|
|
|
= ∑νk |
C vk |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
∂T |
|
|
= ∑νj |
= ∑νk |
|
|
|
= ∆С v |
|||||||||||||||||||||||
|
|
v |
|
j |
∂T |
v |
|
i |
∂T |
v |
|
k |
∂T |
|
|
v |
k |
|||||||||||||||
∂∆H |
|
|
|
|
∂H |
|
|
|
∂H |
|
|
∂H k |
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
= ∑νj |
|
|
− ∑[νi ] |
|
|
= ∑νk |
|
|
|
|
|
|
|
= |
∑νk C pvk = ∆rС p. |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
∂T |
||||||||||||||||||||||||||
|
∂T |
p |
j |
|
∂T p |
i |
|
∂T p |
k |
|
p |
k |
|
|
||||||||||||||||||
∆rС p. = ∆а + ∆bT + ∆cT2 + ∆d T3 + ∆c'T-2.
4.8.5.Анализ уравнения Кирхгоф ф а
4.8.5.1.∆С р = 0 ∆Н T1 = ∆Н T2 . Теплота реакции не зависит от температуры.
4.8.5.2. ∆С р>0 |
T↑ |
∆Н ↑ |
Для эндотермической реакции ∆Н 2 > ∆Н 1 |
|
|
|
|
Для экзотермической реакции ∆Н 2 < ∆Н 1 |
|
4.8.5.3. ∆С р < 0 |
T↑ |
∆Н ↓ |
Для эндотермической реакции ∆Н 2 < ∆Н 1 |
|
|
|
|
Для экзотермической реакции ∆Н 2 > ∆Н 1 |
|
С р |
реагенты |
|
||
|
|
продукты |
|
|
|
|
|
||
|
|
|
|
|
T
∆Н
Э кзотерм ∆С р < 0 ∆С р>0
∆С р = 0 Т 4.8.6. Интегрирование уравнения Кирхгоф ф а
4,8,6,1, Для пересчета теплоты реакции с одной температуры на другую нужно использовать определенный интеграл
2 |
T 2 |
Т |
∫d∆H = ∫∆C p dT . ∆Н Т = ∆Н 298 + |
∫∆Сp dT .= ∆Н 298 + ∆С р (T-298) |
|
1 |
T 1 |
298 |
В небольшом интервале температур ∆С р= const.
Иначе нужно представить ∆С р в виде температурного ряда и только после этого интегрировать уравнение
2 |
T 2 |
|
∫d∆H = ∫∆C p dT = ∫( ∆a + ∆bT + ∆cT 2 + ∆c' T −2 |
)dT |
|
1 |
T 1 |
|
∆Н2 = ∆Н1 +∆a(T2-T1) + ∆b/2 (T22 –T12) + ∆c/3( T23- T13) - ∆c'(1/T2 –1/T1).
4.8.6.2. Для вывода общего уравнения зависимости ∆Н = f (T), нужно брать неопределенный интеграл
∆Н = ∆aT + ∆b/2 T2 + ∆c/3 T3 - ∆c'1/T + ∆Н0
∆Н0 = ∆Н298 –(∆a298 + ∆b/2 2982 + ∆c/3 2983 - ∆c'1/298)
http://www.mitht.org/forum/
14
Лекция № 5
5. Второй закон термодинамики
Первый закон термодинамики устанавливает эквивалентность различных видов энергии и ф орм их передачи. 2-ой закон термодинамики говорит скорее об обратном. Теплота и работа в определенном смысле неэквивалентны – всю работу можно превратить в тепло, а все тепло перевести в работу в периодическом, т.е. циклически повторяющемся процессе нельзя. Все самопроизвольные процессы протекают в одном направлении. Назначение 2-го закона термодинами – установление критериев самопроизвольного протекания разнообразных процессов.
5.1.Классиф икация процессов с точки зрения 2 закона термодинамики.
5.1.1.С а м о п р о и з х в о л ь н ы е процессы выравнивания, протекающие в системе без внешних воздействий, они идут вд установления равновесия.
За счет самопроизвольных процессов может быть получена работа: например, за счет разности давлений можно получить механическую работу или работу электрическуюпьезоэлектричество; за счет разности температур может работать тепловой двигатель или термопара, при установлении химического равновесия можно получить электрическую работу – в гальваническом элементе.
5.1.2.Н е с а м о п р о и з в о л ь н ы е не могут протекать в системе в отсутствие внешних сил
5.1.3.О б р а т и м ы е, р а в н о в е с н ы е, к в а з и с т а т и ч е с к и е или регулируемые процессы , после циклического их завершения не остается никаких изменений ни в системе, ни в окружающей среде.
1) могут изменять направление при бесконечно малом изменении внешних сил
ре ≈ рi ;
2)в прямом и обратном направлении процесс протекает через одни и те же промежуточные состояния – по одному и тому же пути;
3)бесконечно медленно;
4)чаще всего изотермические;
5) |δW| полученная = |δW|затраченная ;
6)δW обр = δW max. > δW необр..
7)Необратимых путей множество, а обратимый только один. Для проведкения
обратимого процесса нужно искусственно тормозить его, прилагая внешнюю силу, преодоление которой позволяет извлекать из системы максимальную работу. Обратимый – идеализированный процесс.
5.2.Формулировки второго закона термодинамики. Закон прошел длительный путь эволюции и сначала был сф ормулирован как основной закон действия тепловых машин
5.2.1.Теорема Карно (1824) “Р азмышление о движущей силе огня” – коэф ф ициент полезного действия обратимого цикла из 2 изотерм и 2 адиабат зависит только от разности температур тепловых резервуаров и не зависит от природы рабочего тела η = (Q 1 – Q 2 )/Q 1 = (T 1 –T 2 )/T 1 = 1 – Т 2 /Т 1 .
5.2.2.Томсон (лорд Кельвин) (1848) ввел понятие абсолютной температуры и (1851) сф оормулировал 2 закон ТД – невозможно построить периодически действующую тепловую машину, которая только бы черпала тепло из одного резервуара и производила механическую работу –невозмоен вечный двигатель 2-го рода
5.2.3.Клаузиус !1850) дал первую ф ормулировку 2 закона ТД – невозможен самопроизвольный переход теплоты от тела менее нагретого к телу более нагретому. Обе ф ормулировки эквивалентны, одна невозможна без другой.
(1854) Ввел понятие энтропии.
5.2.4.Математическое выражение 2 –го з-на ТД
http://www.mitht.org/forum/

15
Для каждой фазы α, содержащей k компонентов, существует экстенсивная ф унк-
ция состояния, называемая энтропией S α = S α(Uα, Vα, n1α, nα2 nαk).
∫dS = 0 , S = ∑S α , изменение которой следующим образом связана с теплотой
|
|
α |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
и температурой процесса |
р |
p1 V 1 T |
1 S 1 |
|
|
|||
|
|
|||||||
dS = |
δQ |
для обратимых процессов; |
|
|
|
|
||
T |
|
|
|
|
||||
|
|
|
|
|
|
|
||
dS > |
δQ |
для необратимых процессов |
|
p2 V 2 T 2 S 2 |
V |
|
||
T |
||||||||
|
|
|||||||
|
|
|
|
|
|
|
||
∆S = S 2 –S 1
Температура интегрирующий делитель, который превратил ф ункцию процесса в изменение ф ункции состояния, а энтропия – тепловая координатаδ
δW = - ∑Y m dX m |
δQ = TdS для обратимых процессов, где Т интенсивный |
m |
|
параметр, а dS изменение экстенсивного свойства, отсюда
d U = TdS – pdV + ∑Y m dX m .
m
5.2.5.Анализ уравнений такого типа привел Каратеодори новой ф ормулировке 2 закона термодинамики, не связанному с теплоыми машинами.
Вблизи любого состояния термически однородной и адиабатически изолированной системы есть бесконечное множество других состояний, не достижи-мых
адиабатическим путем. В этом случае Т становится интегрирующим де-лителем,
аS –ф ункцией состояния.
5.3Р асчет изменения энтропии в арзличных процессах.
Изменение энтропии нельзя непосредственно измерить, измерять можно только теплоту, причем только для обратимых процессов. Количество теплоты может быть различным в обратимом и необратимом процессе, но измененте энтропии в них должно быть одинаковым, если одинаковы начальное и конечное состояния
Q обр. |
|
|
|
δQ |
|
δQ |
|
|
δQ |
|
|
1 |
2 |
dS обр. = |
обр. |
обр. |
> |
необр |
|||||
T |
T |
T |
|||||||||
Q необр. |
|
|
|
|
|
|
|
||||
|
- |
|
|
|
|
|
|
|
|
||
S 2 – S 1 = dS необр. > δTQ необр.
5.3.1. Изменение температуры в закрытой сиситеме |
|
|||||||||||||||||||||||||||||||
5.3.1.1. V = const |
dS = |
|
δQ |
|
|
= |
dU |
= |
|
nC V |
|
dT |
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
T |
|
|
|
|
T |
|
|
|
|
T |
|
|
|||||||||||||||||||
|
|
2 nC V |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
2 |
|
|
|
|
|
|
2 |
|
dT |
|
|
|
|
|
|
|
|
|
T |
|
|
|
|
|
|
|||||||
∫ |
dS = |
∫ |
|
|
|
dT |
= n C V |
∫ |
|
T |
|
= nC V ln |
T |
1 |
, если С V = const, т.е. интервал темпера- |
|||||||||||||||||
T |
|
|
|
|||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
1 |
|
1 |
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
тур небольшой. |
|
|
|
|
|
|
|
|
nC p |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
2 |
|
2 |
|
|
|
|
= n ∫ a + bT + cT |
2 |
||||||||||||||
5.3.1.2. p = const |
|
∫dS = ∫ |
|
|
dT |
dT = |
||||||||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||||||
|
|
|
|
T |
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
1 |
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
T |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
= n[ aln |
|
T 2 |
|
+ b(T 2 |
|
− T 1 )+ |
c |
(T 2 |
2 − T 1 |
2 )]. |
|
|
|
|
|
|
|
|
||||||||||||||
|
T 1 |
|
|
2 |
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
http://www.mitht.org/forum/

16
∆SV = 5 кал/(моль*К) * ln 373K/273 = 1,56 кал/(моль*К) = 6,53 Дж/(моль*К) С и S имеют одинаковую размерность.
5.3.2. Изотермическое расширение-сжатие идеального газа
|
dU + pdV |
|
pdV |
|
nR dV |
2 |
nRdV |
|
|
|
|
|
V2 |
|
p1 |
|
|||||||
dS = |
|
|
= |
|
= |
|
|
|
; ∆S = ∫ |
|
|
|
|
= nRln |
|
= nRln |
|
. |
|||||
T |
|
T |
V |
|
V |
V1 |
p2 |
||||||||||||||||
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|||||||||||
5.3.3. Смешение газов |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
na V a |
nbV b |
|
|
|
|
|
|
nb na ,, V a+b, p, T |
|
|
|
|
|
|
nk/ Σnk = Nk |
||||||||
pT |
pT |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
мольная доля |
||
|
|
|
|
V a + V b |
|
|
|
|
V a + V b |
|
|
||||||||||||
∆S = ∆S a + ∆S b = na R ln |
+ nb R ln |
|
= |
|
|
|
|
||||||||||||||||
V a |
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
V b |
|
|
|
|
|
|
|
||||
|
|
= na |
R ln |
n a |
+ n b |
|
+ nb R ln |
|
n a |
|
+ n b |
|
= -R (na ln Na + nblnNb). |
||||||||||
|
|
|
n a |
|
|
n b |
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Изменение энтропии при смешении газов зависит от концентрации получаемой смеси 5.3.4. Фазовые переходы
dS = |
∆H ф.п. |
|
dn ∆S = |
∆H ф.п. |
|
. |
||
|
|
|
|
|
|
|||
T ф.п. |
|
T ф.п. |
|
|||||
|
|
|
|
|
||||
5.3.5. Химические реакции
∆S = ΣνjS j - Σ νi S i = ΣνkS k.
5.4.Э нтропия – критерий направления самопроизвольных процессов в изолированной системе
dS > 0 самопроизвольный процесс
dS = 0 равновесие; в стабильном равновесии изолированная система достигает
максимума энтропии d2S < 0
В неизолированной системе энтропия может уменьшатьсяБ но при этом должен происходить рост энтропии в окружающей среде
Процесс |
∆S системы |
∆S окружающей среды |
∆S общ. |
Циклический обратимый |
0 |
0 |
0 |
|
|
|
|
Нециклический обратимый |
>0 |
<0 |
0 |
- « - |
< 0 |
> 0 |
0 |
Циклический необратимый |
0 |
>0 |
>0 |
Нециклич. необратимый |
>0 |
<0 |
>0 |
- « - |
<0 |
>0 |
>0 |
http://www.mitht.org/forum/

Лекция № 6
5.5. Статистический смысл энтропии
Термодинамические соотношения не зависят от природы веществ, но термодинамические свойства индивидуальных соединений находят опытным путем. Задача статистической термодинамики – определение характеристик веществ на основании свойств образующих их частиц, законов их движения и взаимодействия. Статистическая термодинамика начала развиваться с конца XIX века. Основные принципы ее были разработаны в трудах Больцмана, Гиббса, Планка, Эйнштейна, Эренфеста, Т. Афанасьевой-Эренфест.
5.5.1. Макро- и микросостояния термодинамической системы. М а к р о состояние системы задается набором термодинамических параметров, которые имеют смысл для всей системы в целом.
М и к р о состояние – это состояние отдельных частиц системы, которые можно разделить на квантовые и классические. Критерием отнесения может служить соотношение разности
энергетических уровней ∆ε и энергией теплового кванта kT: если ∆ε>> kT, то соответствующая степень свободы относится к квантовым характеристикам, в противном случае – к классическим. Будем полагать, что квантовые характеристики постоянны и классическое движение ограничивается поступательным. Тогда для задания состояния каждой частицы необходимо задать для каждой частицы 3 координаты – хk, yk, zk и 3
проекции импульса рхk, руk |
и рzk, т.е. всего 6 N переменных, где k 1,2, 3. N. |
(Е.А.П. Примечание: |
Квантор здесь ошибочный !) |
N очень велико для термодинамических систем, и это приводит к новым статистическим закономерностям, которые нельзя свести к механическим. Характерной чертой их является представление о динамических переменных как случайных величинах, которым можно приписать определенную вероятность.
5.5.2. Фазовое пространство. Наглядно состояние системы можно представить, используя понятие фазового пространства. Ф а з о в о е пространство – математическое понятие, это условное пространство возможных микросостояний системы. Когда Вы изображали электронные конфигурации атомов в виде клеточек, заполненных электронами, то имели дело с фазовым пространством электронов в атоме. В нашем случае координатами фазового пространства служат координаты и проекции импульсов частиц, т.е. это пространство 6N-мерное, микросостояние всей системы изображается точкой в этом пространстве, при изменении состояния изображающая точка описывает траекторию в фазовом пространстве. Такое пространство обозначают как Г-пространство. Но молекулы одного вещества обладают одинаковыми свойствами, поэтому можно свести
описание микросостояния системы к рассмотрению 6-мерного µ- пространства
Фазовое пространство Размерность Описание микросост. системы Изменение сост.
Г |
6N |
Точка |
Линия |
µ |
6 |
Распределение N точек |
Измен. распред. |
Изменение координат и импульсов происходит непрерывно, поэтому кажется, что состояний бесконечное множество, и сосчитать их очень трудно. Так было во времена Гиббса, в последней четверти ХIХ в. Однако развитие квантовой теории сделало фазовое пространство счетным, когда Гейзенбергом был сформулирован принцип неопределенности, который позволяет разбить фазовое пространство на счетное количество фазовых ячеек, каждая из которых отвечает различимому микросостоянию частицы:
∆х ∆рх>h для одномерного движения
∆х ∆рх ∆у ∆ру ∆z ∆рz >h3 для движения в трехмерном пространстве. рх
http://www.mitht.org/forum/

х
5.5.3.Термодинамическая вероятность.
5.5.3.1.В равновесии термодинамические параметры системы постоянны , но микро-характеристики молекул претерпевают непрерывные изменения. Таким образом, макросостояния системы реализуется множеством микросостояний.
Число микросостояний, совместимых с данным макросостоянием, называется термодинамической вероятностью
5.5.3.2.Принципы расчета термодинамической вероятности.
5.5.3.2.1.Микросостояния отличаются друг от друга перестановкой частиц.
5.5.3.2.2.Перестановки внутри одной ячейки не ведут к новому микросостоянию.
5.5.3.2.3.В одно макросостояние входят все микросостояния с одинаковым количественным распределением частиц по ячейкам, т.е. задать макросостояние на микроуровне можно определением числа частиц в каждой ячейке фазового пространства Ni/
5.5.3.2.4.Основной постулат – для изолированной системы (U= const) все
микросостояния имеют одинаковую вероятность. Функция распределения ρ = const. Это позволяет легко рассчитывать термодинамическую вероятность состояния системы, состоящей из N частиц, распределенных по s ячеек фазового пространства, используя формулу комбинаторики для числа перестановок:
|
|
|
|
|
|
N! |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
W = |
|
|
|
|
|
|
|
|
|
, а общее число микросостояний = S / |
||||||||||||
N !N |
2 |
!N |
3 |
!...N |
s |
! |
||||||||||||||||
|
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Пример: 2 частицы в двух ячейках (микросостояниях), N = 2, S = 2. |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
1 2 |
|
|
2 |
|
1 |
|
|
|
||||
4микросостояния |
|
|
|
1 |
|
2 |
1 2 |
|
|
|||||||||||||
разбиты на 3 макросостояния: |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
N1 |
|
|
N2 |
|
W |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
1 |
|
2 |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
2 |
|
1 |
|
|
||
5.5.3.3. Наибольшая |
|
2 |
|
|
|
|
0 |
|
1 |
|
|
|||||||||||
|
термодинамическая вероятность получается при равномерном |
|||||||||||||||||||||
распределении N частиц по ячейкам с одинаковой энергией. |
||||||||||||||||||||||
W |
= |
|
|
|
N! |
|
|
|
. С увеличением числа частиц термодинамическая вероятность |
|||||||||||||
|
|
|
|
|
|
|
||||||||||||||||
max |
|
[(N / S)!]s |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
равномерного распределения частиц по ячейкам очень сильно возрастает |
||||||||||||||||||||||
N |
|
|
S |
|
|
|
|
|
Wmax |
|
|
|
|
|
|
|
||||||
2 |
|
|
2 |
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
||
4 |
|
|
2 |
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
|
|
|
||
6 |
|
|
3 |
|
|
|
|
15 |
|
|
|
|
|
|
|
|
|
|
||||
15 |
|
|
3 |
|
|
|
|
756756 |
|
|
|
|
|
|
|
|||||||
5.5.3.4. Направление самопроизвольных процессов. Изолированная термодинамическая система стремится к состоянию с наибольшей термодинамической вероятностью, которое достигается при установлении равновесия.
5.5.4. Связь энтропии с термодинамической вероятностью предложена на основании свойств энтропии и термодинамической вероятности
Аддитивность S = f(W); |
S = S1 + S2 . |
Мультипликативность |
W = W1*W2. |
S(W) = S(W1*W2) = S1(W1) + S2 (W2) S = k ln W,
Где k – постоянная Больцмана
Таким образом, энтропия характеризует неупорядоченность системы, ее беспорядок.
http://www.mitht.org/forum/
5.5.5. Процессы , ведущие к увеличению энтропии
5.5.5.1.Увеличение числа частиц, например, распад молекул, диссоциация.
5.5.5.2.Увеличение объема фазового пространства, т.е. увеличение числа ячеек, или возможных состояний.
А) увеличение объема, расширение; смешение, свертывание полимера в клубок; Б) увеличение числа доступных уровней энергии, густота энергетического
спектра, уменьшение расстояния между уровнями. Это связано с характером движения. Наиболее беспорядочно поступательное движение, затем следует вращательное, колебательное.
5.5.6. Распределение молекул по энергии при постоянной температуре.
Система и термостат образуют изолированную систему, но сама система мала по сравнению с термостатом. В этих условиях частицы будут обладать разной энергией в состоянии равновесия.
N1 |
ε1 |
ΣNi = N = const; ΣεiNi = U = const |
N2 |
ε2 |
|
Nm……………εm…… |
|
|
Распределение |
Больцмана: |
Ni |
= Agi exp(−εi / kT); где |
gi–кратность вырождения |
||||||
N |
||||||||||
|
|
|
|
|
|
|
|
|
||
уровня, |
|
т.е. |
число состояний |
с заданным |
i- уровнем энергии |
|||||
∑ |
Ni |
= |
1 = |
i=∞ |
|
|
|
i=∞ |
|
|
N |
∑Agi exp(−εi / kT);A =1/ |
∑gi exp(−εi / kT) =1/ Q; |
||||||||
i |
|
|
i=1 |
|
|
|
i=1 |
|
||
Q – статистическая сумма, характеристика, которая связывает свойства частиц (набор их допустимых энергетических уровней и кратность их вырождения) с макроскопическим параметром – температурой. Поэтому именно эта величина будет фигурировать далее во всех формулах статистической термодинамики.
i=∞ |
Ni |
= gi exp(−εi / kT) / Q; |
Q= ∑gi exp(−εi / kT) ; |
N |
|
i=1 |
|
6. Третий закон термодинамики
6.1.Формулировки
6.1.1.Тепловая теорема Нернста (1906 г.)
При температуре, близкой к абсолютному нулю, многие свойства системы становятся независимыми от температуры, т.е. становятся постоянными, например теплоты химических реакций, объем системы, поверхностное натяжение и др.
lim d∆dTr H = 0 ∆Cp = 0 . Теплоемкости всех веществ становятся одинаковыми и
стремятся к 0. Изменение энтропии при всех процессах при абсолютном нуле равно
0 6.1.2. Постулат Планка
Энтропия идеального кристаллического тела при абсолютном нуле равна 0. статистическое обоснование – все частицы находятся на низшем энергетическом уровне, т.е. занимают 1 ячейку фазового пространства. Термодинамическая вероятность такого состояния равна 1, а энтропия = 0.
6.1.3. Недостижимость абсолютного нуля . Охлаждение достигается за счет
адиабатического расширения (∆Т < 0) и изотермического сжатия (∆S < 0) , при последнем процессе нужно отводить тепло, что возможно только к менее нагретому телу. При абсолютном нуле невозможно понизить энтропию, поэтому абсолютный нуль недостижим, хотя в настоящее время достигнуты температуры в несколько сотых градуса Кельвина.
http://www.mitht.org/forum/
(Е.А.П. Примечание: В настоящее время известны способы перехода в область отрицательной температуры. Однако абсолютное значение нега-температуры не превышает нескольких сотых градуса. Смена знака температуры достигается методом адиабатического размагничивания... Смысл понижения температуры статистический, и он состоит в инверсии заселённостей спиновых уровней ядер...
Эффективно применяется в этом процессе лазерное излучение.) 6.2.Остаточная энтропия
Реальные кристаллы могут иметь дефекты – дырки по Шоттки и Френкелю,
(Е.А.П. Примечание: Немного перепутано – тв. Тело с жидкостью)
дислокации, нарушения кристаллической решетки, основной уровень может быть вырожденным. В этих случаях энтропия при абсолютном нуле будет иметь значение, отличное от 0, которое называется остаточной энтропией. Ее можно оценить статистическим методом по формуле S = R ln g0
6.3.Абсолютная энтропия – это изменение энтропии, отсчитанная от абсолютного нуля до данного состояния. Для ее расчета необходимо суммировать изменение энтропии во всех процессах нагревания и фазовых переходах:
0 |
|
Нагревание твердого тела |
Нагревание жидкости |
Нагревание газа |
||
Недоступен для измерения |
Плавление |
Кипение |
||||
Определяется по Дебаю |
|
|
|
|
||
http://www.mitht.org/forum/

21
Лекция № 7
7.Применение 2-го закона термодинамики к изотермическим процессам
7.1.Фундаментальное уравнение Гиббса
Уравнение 2-го закона не очень удобно для использования, так как в нем характеристики системы выражаются через ф ункции процесса. Нужно, чтобы свойства системы выражались через ее параметры.
TdS ≥dU +pdV + δW’, для закрытой системы и δW’=0
dS = 1/T dU + p/T dV |
|
∂S |
|
1 |
|
∂S |
|
p |
|
||
|
|
|
= |
|
, |
|
|
= |
|
; S = f (U,V). |
|
|
|
|
|
||||||||
|
|
∂U V |
|
T |
|
∂V U |
|
T |
|
||
Э нтропия системы является ф ункцией от внутренней энергии и объема , производные по этим параметрам выражаются через отношение интенсивных характеристик системы, направление самопроизвольных процессов в звкрытых системах выражается
неравенством dS U,V > 0.
Перепишем это уравнение для внутренней энергии
dU = TdS – pdV |
|
оно содержит эквивалентную инф ормацию о системе и называется ф |
||||
|
|
|
у н д а м е н т а л ь н о е у р а в н е н и е |
|||
|
|
|
||||
|
|
|
Гиббса и объединяет 1-ый и 2-ой законы ТД. |
|||
U = f (S ,V); |
|
∂U |
|
∂U |
= p. |
|
|
|
|
= T ; |
|
||
|
|
∂S V |
|
∂V S |
|
|
dU ≤ TdS - pdV - δW’. Направление самопроизвольных процессов может быть выражено через изменение внутренней энергии: dU S ,V < 0. Такая ситуация имеет место в механических системах, в которых энтропия постоянна. Устойчивое равновесие достигается при минимуме потенциальной (внутренней) энергии:
dU S ,V S ,V < 0.
7.2. Критерии направления самопроизвольных процессах в изотермических условиях. Термодинамические потенциалы.
7.2.1. Э нергия Гельмгольца. 7.2.1.1. Физический смысл
dU ≤ TdS - pdV - δW’ = TdS - δW. Преобразуем это уравнение таким образом, чтобы ф ункции состояния попали в одну сторону неравенства – влево: dU - TdS ≤ -δW.
Представим изотермический процесс и проинтегрируем это уравнение
2 |
2 |
|
|
|
|
∫dU − ∫TdS |
≤ -W T ∆U - T∆S ≤ -W T U 2 – U 1 – T(S 2 – S 1 ) ≤ -W T . |
||||
1 |
1 |
|
|
|
|
(U 2 –TS 2 ) – (U 1 –TS 1 ) ≤ - W T . |
Получили новую ф ункцию состояния |
||||
∫dA = 0 ; ∆A = A2 – A1 |
|
|
|
||
A ≡ U – TS ; |
∆A ≤ -W T ; |
-∆A ≥ W T ; |
-∆A= (W T )max |
|
|
Убыль энергии Гельмгольца равна максимальной работе обратимого изотермического процесса. В необратимом процессе работа полученная оказывается меньше убыли энергии Гельмгольца, а затраченная забота больше роста энергии Гельмгольца.
7.2.1.2. Направление самопроизвольных процессов при Т,V = const. Напишем полный диф ф еренциал энергии Гельмгольца и подставим в него ф ундаментальное уравнение Гиббса dU ≤ TdS - pdV - δW’
http://www.mitht.org/forum/
22
dA = dU – TdS – S dT dA ≤ TdS - pdV - δW’ – TdS – S dT dA ≤ - S dT - pdV - δW’ .
При Т,V = const в самопроизвольном процессе δW’≥0, отсюда Критерием направления самопроизвольного процесса будет уменьшение энергии Гельмгольца:
dA V,T ≤ 0 условие направление самопроизвольного процесса при Т,V = const. dA V,T = 0; d 2A V,T > 0 – условие стабильного равновесия при Т,V = const.
С табильное равновесие достигается при минимуме энергии Гельмгольца. 7.2.1.3. Полный диф ф еренциал энергии Гельмгольца в звкрытой системе dA = - S dT – pdV
|
∂A |
|
∂A |
= −p. С ростом температуры и объема энергия Гельмгольца |
|
|
= −S; |
|
|
|
∂T V |
|
∂V T |
|
всегда падает, так как энтропия и давление системы всегда имеют положительное значение, и производная будет отрицательной. 7.2.2. Э нергия Гиббса.
7.2.2.1. Физический смысл
Проинтегрируем ф ундаментальное уравнение Гиббса при T,p = const dU - TdS + pdV ≤ - δW’
d(U –TS +P V) ≤ - δW’
∆(U –TS +P V) ≤ - W’p,T Новая ф ункция состояния – энергия Гиббса
∫dG = 0 , ∆G = G 2 – G 1 . |
|
|
|
|
Убыль энергии |
G ≡ U + pV– TS ; ∆G ≤ -W’p,T ; |
-∆G ≥ W’p,T ; -∆G = (W’p,T )max |
|
|
|
Гиббса равнв |
|
|
максимальной полезной работе изобарно-изотермического процесса. 7.2.2.2. Критерии направления самопроизвольного процесса при T,p = const
dG p,T ≤ 0 ∆G p,T ≤ 0 |
Процессы при T,p = const идут в сторону убыли энергии |
|
Гиббса. |
|
С табильное равновесие достигается при минимуме |
|
|
|
dG p,T = 0 ; ∆G p,T = 0 |
|
энергии Гиббса |
d 2G p,T ≥ 0 |
|
|
7.2.2.3. Полный диф ф еренциал энергии Гиббса в закрытой системе dG = dU + pdV + Vdp – TdS – S dT из определения ф ункции. Подставим ф ундаментальное уравнение Г иббса
dG = TdS - pdV + pdV + Vdp – TdS – S dT = Vdp –S dT. Определяющими параметрами для энергии Гиббса будут – р и Т, G = f(p,T)
dG = Vdp –S dT
∂G |
|
|
∂G |
= V . |
|
|
|
|
= −S; |
|
|
|
∂T |
p |
|
∂p T |
|
7.3.Характеристические функции
7.3.1.Определение
Характеристические ф ункции - это экстенсивные ф ункции состояния си-стемы, посредством которых, а также производных от них различных поряд-ков по соответствующим параметрам, выражаются в я в н о м виде все свойства системы. Э нтропия не является характеристической ф ункцией. Каждая ф ункция состояния связана со своим набором переменных:
Мнемоническое правило P V
http://www.mitht.org/forum/

|
|
|
|
|
|
|
|
|
|
|
|
|
|
23 |
|
|
S |
|
T |
|
|
|
|
|
|
|
|
|
|
|
U |
H |
|
A |
G |
|
|
|
|
|
|
|
|
|
Можно написать внутреннюю энергию как ф ункцию T , V |
|
|
|
|
||||||||||
U = f ( T,V) |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
∂U |
|
∂U |
dV |
Не определены энтропия и давление - |
|
|||||||
dU = |
dT + |
|
|
|||||||||||
|
|
∂T V |
|
∂V T |
|
|
|
|
|
|
|
|
|
|
|
|
C V |
0 |
|
важнейшие характеристики системы |
|
|
|
||||||
|
|
( Для идеальных газов) |
|
|
|
|
|
|
|
|
||||
7.3.2. Таблица характеристических ф ункций |
|
|
|
|
|
|
|
|
||||||
Функ- |
Параметры |
|
Условие |
Условие |
|
|
Производные |
|
|
Физиче- |
||||
ция |
связан- |
самопроиз- |
равновесия |
|
|
|
ф ункции |
|
|
|
ский |
|||
|
ные с ней |
|
вольных |
|
|
|
|
|
|
|
|
смысл |
||
|
|
|
процессов |
|
|
|
|
|
|
|
|
|
||
U |
|
S ,V |
|
∆U S ,V <0 |
∆U S ,V =0 |
|
∂U |
∂U |
|
= −p |
|
∆U= QV |
||
|
|
|
|
|
|
|
|
|
|
= Т; |
|
|
|
|
|
|
|
|
|
|
|
|
|
∂S V |
∂V |
S |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
H |
|
S,H |
|
∆HS,p<0 |
∆HS,p=0 |
|
∂H |
∂H |
= V |
|
∆H = Qp |
|||
|
|
|
|
|
|
|
|
|
= T; |
|
|
|
||
|
|
|
|
|
|
|
|
∂S p |
∂p |
S |
|
|
|
|
A |
|
T,V |
|
∆AT,V<0 |
∆AT,V=0 |
|
∂A |
∂A |
= −p. |
|
-∆A = |
|||
|
|
|
|
|
|
|
|
|
|
= −S; |
|
|
(WT)max |
|
|
|
|
|
|
|
|
|
∂T V |
∂V T |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
||
G |
|
T,p |
|
∆G p,T<0 |
∆G p,T=0 |
|
∂G |
∂G |
|
= V. |
|
-∆G = |
||
|
|
|
|
|
|
|
|
|
|
= −S; |
|
|
(W’pT)max |
|
|
|
|
|
|
|
|
|
|
∂T p |
∂p |
T |
|
|
|
7.3.3. Полные дифференциалы и частные производные характеристических |
||||||||||||||
ф ункций в закрытых системах |
|
|
|
|
|
|
|
|
|
|||||
dU = TdS – pdV |
|
|
Э ти выражения получились как преобразования dН = |
|||||||||||
TdS – Vdp |
|
ф ундаментального уравнения Гиббса к другим |
|
|||||||||||
dA = - S dT – pdV |
|
|
переменным, но содердат эквивалентную ему |
|||||||||||
dG = Vdp –S dT |
|
|
инф ормацию |
|
|
|
|
|
|
|
|
|||
В этих выражениях термодинамические параметры получили свое определение, независимое от эмпирической основы. Производные от характеристических ф ункций по экстенсивным параметрам дают интенсивные характеристики системы и, наоборот, производные по интенсивным переменным приводят к экстенсивным параметрам. В ф ундаментальном уравнении Гиббса естественными параметрами служат экстенсивные величины - энтропия и объем. Э то очень неудобно для практических целей, так как нет способа поддерживать постоянную энтропию в ТДС . В то же время интенсивные параметры легко контролировать в различных процессах, поскольку они выравниваются автоматически в состоянии равновесия. Поэтому наиболее важной ф ункцией для химиков является энергия Гиббса, для которой
определяющими параметрами служат температура и давление. При постоянстве этих параметров проходит большинство химических процессов. Поэтому остановимся более подробно на анализе этой ф ункции 7.3.4. Зависимость энергии Гиббса от температуры и давления
G ≡ U + pV– TS ≡ H -TS
∆G pT ≡ ∆ H -T∆S ∆ rG pT ≡ ∆ rH -T∆rS
http://www.mitht.org/forum/
24
∆ H характеризует общееи изменение энергии системы в ходе процесса, а изменение энергии Гиббса показывает, какая часть ее может быть превращена в полезную работу, поэтому раньше ее называли свободной энергией, в противоположность энтропийному члену, который характеризовал связанную энергию, недоступную для превращения в работу.
|
∂G |
= V. С ростом давления энергия Гиббса увеличивается, так как |
|
|
|
|
∂p T |
|
V> 0, но темп роста замедляется, так как объем постепенно уменьшается, Причем для разных идеальных газов эти кривые будут одинаковыми
G
T = const |
G |
p = const |
∂G |
|
p |
T |
||
= −S. С ростом температуры энергия Гиббса убывает, так как энтропия |
|||||
|
∂T |
|
|||
|
p |
|
|
||
всегда положительна, причем падение энергии Гиббса будет ускоряться из-за роста энтропии при поъеме температуры.
7.3.5. Уравнение Гиббса – Гельмгольца |
|
|
|
|
|
|||||||||||||||||||||
Представим себе химическую реакцию А = B |
|
|
||||||||||||||||||||||||
∂G |
A |
|
|
|
|
|
∂G B |
|
|
|
|
∂∆ |
|
G |
|
|
||||||||||
|
|
|
|
|
|
= −S A . |
|
|
|
|
|
|
|
|
|
= −S B . |
|
|
r |
|
|
= −∆r S. |
||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
∂T |
|
|
|
|
|
|
|
∂T |
p |
|
||||||
∂T |
|
|
p |
|
|
|
|
p |
|
|||||||||||||||||
∆ rG |
|
|
|
|
|
|
∂∆ |
|
G |
|
|
|
|
уравнение Гиббса-Гельмгольца. |
||||||||||||
pT |
≡ ∆ rH +T |
|
r |
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
∂T |
|
p |
|
|
|
|
|
|
|
|
|
|
|
||||
В уравнении Гиббса –Гельмгольца мы избавились от неприятной величины – изменения энтропии в химической реакции и заменили ее температурной зависимостью измеряемой величины – изменением энергии Гиббса в химической реакции, которая связана с измеряемой характеристикой – полезной работой обратимого изотермическлгл процесса.
Лекция № 8
8. Фундаментальное уравнение Гиббса для открытых систем
8.1. Дифференциалы характеристических функций
dU = TdS – pdV - δW’ для открытых систем нужно учесть химическую работу
δW хим. = - ∑µk dn k |
т.е. обмен веществом с окружающей средой. |
k |
|
http://www.mitht.org/forum/

25
dU = TdS – pdV + ∑µk dn k . |
U = f ( S ,V,nk) |
k |
|
dН = TdS + Vdp + ∑µk dn k . |
H = f S ,p, nk) |
k |
|
dA = - S dT – pdV + ∑µk dn k . |
A = f (V,T, nk) |
k |
|
dG = Vdp –S dT + ∑µk dn k . |
G = f (p,T, nk) |
k |
|
Гиббс ввел в качестве переменной массу, а точнее количество молей компонентов
8.2.Понятие химического потенциала.
µkα - химический потенциал всегда является интенсивной характеристикой
состояния компонента k в ф азе α.
Химический потенциал k –компонента в ф азе α показывает изменение любой характеристической ф ункции при введении в систему (ф азу) 1 моля k-вещества при постоянных естественных параметрах данной характеристической ф ункции и неизменном количестве других компонентов ( или при неизменном составе ф азы).
|
|
∂U |
|
|
∂H |
|
|
∂A |
|
|
∂G |
|
|
|
µk |
|
|
|
|
|
|
|
|
. |
|||||
|
|
|
|
|||||||||||
= |
|
|
= |
|
|
= |
|
|
= |
|
|
|||
|
|
∂n k S ,V ,nl |
|
∂n k S,p,nl |
|
∂n k T ,V ,nl |
|
∂n k p,T ,nl |
|
|||||
Наиболее часто используется именно последнее выражение химического потенциала через энергию Гиббса.
8.3. Интегрирование фундаментального уравнения Гиббса по числу молей при постоянных температуре, давлении и составе. Будем увеличивать число молей в
системе от нуля до n. В фундаментальном уравнении в качестве независимых
переменных выступают экстенсивные величины, которые зависят от числа молей. Если n =0, то и все экстенсивные переменные имеют в качестве нижнего предела 0.
U S V
∫dU = T ∫dS – p ∫dV dV +
0 0 0
n
∑µk ∫ dn k .
k 0
U = TS – pV + Σµknk |
G = Σµknk. Если система однокомпонентная, |
H = U + pV = TS + Σµknk |
то уравнение вовсе упрощается G = µn. |
A = U –TS = -pV + Σµknk |
Отсюда вытекает важное следствие: |
G = U + pV – TS = Σµknk |
|
µ = G/n = G Химический потенциал чистого вещества равен мольной энергии
Гиббса.
8.4. Уравнение Гиббса-Дюгема
Напишем полный дифференциал внутренней энергии из определения
dU = TdS + SdT – pdV – Vdp + ∑µk dn k + ∑n k dµk .
k k
Подставим в него фундаментальное уравнение Гиббса:
TdS – pdV + ∑µk dn k = TdS + SdT – pdV – Vdp + ∑µk dn k + ∑n k dµk .
k |
k |
k |
|
Уравнение Гиббса-Дюгема |
|
0 = SdT – Vdp + ∑n k dµk + ΣXidYi |
||
k |
|
|
Независимыми переменными в уравнении Гиббса-Дюгема служат интенсивные
характеристики. Оно показывает, что изменение интенсивных
http://www.mitht.org/forum/

26
параметров смистемы связано между собой и изменение одного интенсивного параметра влечет за собой изменение другого. Изменяете температуру – изменяются давление и химические потенциалы компонентов, и т.д. Таким образом для задания состояния системы нельзя использовать одни интенсивные параметры, нужно задать хотя бы один экстенсивный.
Е сли T,p = const, то уравнение Гиббса Дюгема приобретает вид
∑n k dµk = 0 . Э то означает, что химические потенциалы веществ в системе,
k
связаны между собой. Э то уравнение играет большую роль в теории растворов. 8.5. Выражение химического потенциала компонента идеального газа
8.5.1. Чистое вещество в закрытой системе (n = const) µ = G/n = G .
dG = - S dT + Vdp, при постоянной температуре dG T = Vdp.
Проинтегрируем последнее выражение от некоторого стандартного состояния до заданного с учетом уравнения Менделеева-Клапейрона
V = R T/p.
G |
p |
dp |
|
|
p |
|
|
|
∫dG = R T ∫ |
= R T ln |
|
|
|||||
p |
0 |
|||||||
G 0 |
p0 |
|
|
p |
||||
GT= G0T + RT ln p/p0. |
µT = µ0T + RT ln~p |
Обе записи эквивалентны. |
||||||
|
|
|
|
|
|
|
||
Так как p0 = 1 атм, то его часто опускают, но надо помнить, что под логарифмом не может стоять размерная величина, а только относительное давление ( или приведенное к стандартному давлению). При постоянной температуре
химический потенциал пропорционален логарифму приведенного давления, а
µ0T - это стандартный химический потенциал идеального газа при заданной
температуре ( и давлении = 1 атм)
8.5.2. Смесь газов Рассмотрим процесс смешения: исходное состояние – 2 газа в стандартных
условиях, конечное состояние смесь газов при той же температуре и том же
|
|
|
|
|
общем давлении. |
|
|||||
|
p0A, T, nA |
p0B, T, nB |
|
||||||||
|
|
|
|
|
|
|
|||||
|
pA ++ pB = p0, T, nA,nB |
|
|
|
|
||||||
|
p |
p |
RT |
|
|
|
|
pA |
0 |
|
|
∆G A = ∫ Vdp = n ∫ |
p |
dp = nRT ln |
|
; |
∆GB = nBRT ln pB/pB |
. |
|||||
0 |
|||||||||||
|
p0 |
p0 |
|
|
|
|
pA |
|
|
||
∆G = ∆G A + ∆GB |
= RT(nA ln pA/pA0 + nB ln pB/pB0) |
|
|||||||||
∆GpT = nA(µA - µA |
0) + nB |
(µA - µA0). Отсюда |
|
||||||||
µA = µA0 + RT ln pA/pA |
0 и µB = µB0 + RT ln pB/pB0, т.е. химический потенциал |
||||||||||
компонента идеальной газовой смеси зависит от парциального давления этого компонента µk = µk0 + RT ln ~pk/
http://www.mitht.org/forum/

25
Лекция № 8
8. Фундаментальное уравнение Гиббса для открытых систем
8.1. Диф ф еренциалы характеристических ф ункций
dU = TdS – pdV - δW’ для открытых систем нужно учесть химическую работу
δW хим. = - ∑µk dn k |
т.е. обмен веществом с окружающей средой. |
|
k |
|
|
dU = TdS – pdV + ∑µk dn k . |
U = f ( S , V, nk) |
|
k |
|
|
dН = TdS + Vdp + ∑µk dn k . |
H = f ( S , p, nk) |
|
k |
|
|
dA = - S dT – pdV + ∑µk dn k . |
A = f (V, T, nk) |
|
|
k |
|
dG = Vdp –S dT + ∑µk dn k . |
G = f (p, T, nk) |
|
k |
|
|
Гиббс ввел в качестве переменной массу, а точнее количество молей компонентов
8.2.Понятие химического потенциала.
µkα - химический потенциал всегда является интенсивной характеристикой
состояния компонента k в ф азе α.
Химический потенциал k –компонента в ф азе α показывает изменение любой характеристической ф ункции при введении в систему (ф азу) 1 моля k-вещества при
постоянных |
|
естественных |
параметрах |
данной |
характеристической ф ункции и |
|||||||||
неизменном количестве других компонентов ( или при неизменном составе ф азы). |
||||||||||||||
|
|
∂U |
|
|
∂H |
|
|
∂A |
|
|
|
∂G |
|
|
µk = |
|
|
|
|
|
|
|
|
|
. |
||||
|
|
|
|
|
||||||||||
|
|
|
= |
|
|
= |
|
|
|
= |
|
|
||
|
|
∂n k S ,V ,nl |
|
∂n k S,p,nl |
|
∂n k T ,V ,nl |
|
∂n k p,T ,nl |
|
|||||
Наиболее часто используется именно последнее выражение химического потенциала
через энергию Гиббса.
8.3. Интегрирование ф ундаментального уравнения Гиббса по числу молей при постоянных температуре, давлении и составе. Будем увеличивать число молей в системе от нуля до n. В ф ундаментальном уравнении в качестве независимых переменных выступают экстенсивные величины, которые зависят от числа молей. Е сли n =0, то и все экстенсивные переменные имеют в качестве нижнего предела 0.
U |
S |
V |
∫dU |
= T ∫dS – p ∫dV dV + |
|
0 |
0 |
0 |
n
∑µk ∫ dn k .
k 0
U = TS – pV + Σµknk |
G = Σµknk. Е сли система однокомпонентная, |
H = U + pV = TS + Σµknk |
то уравнение вовсе упрощается G = µn. |
A = U –TS = -pV + Σµknk |
Отсюда вытекает важное следствие: |
G = U + pV – TS = Σµknk |
|
µ = G /n = G Химический потенциал чистого вещества равен мольной энергии Гиббса.
8.4. Уравнение Гиббса-Дюгема Напишем полный диф ф еренциал внутренней энергии из определения
dU = TdS + S dT – pdV – Vdp + ∑µk dn k + ∑n k dµk .
k |
k |
Подставим в него ф ундаментальное уравнение Гиббса:
TdS – pdV + ∑µk dn k = TdS + S dT – pdV – Vdp + ∑µk dn k + ∑n k dµk .
k |
k |
k |
http://www.mitht.org/forum/

26
|
Уравнение Гиббса-Дюгема |
0 = SdT – Vdp + ∑n k dµk + ΣXidYi |
|
k |
|
Независимыми переменными в уравнении Гиббса-Дюгема служат интенсивные характеристики. Оно показывает, что изменение интенсивных параметров смистемы связано между собой и изменение одного интенсивного
параметра влечет за собой изменение другого. Изменяете температуру – изменяются давление и химические потенциалы компонентов, и т.д. Таким образом для задания состояния системы нельзя использовать одни интенсивные параметры, нужно задать хотя бы один экстенсивный.
Е сли T,p = const, то уравнение Гиббса Дюгема приобретает вид
∑n k dµk = 0 . Э то означает, что химические потенциалы веществ в системе,
k
связаны между собой. Э то уравнение играет большую роль в теории растворов. 8.5. Выражение химического потенциала компонента идеального газа
8.5.1. Чистое вещество в закрытой системе (n = const) µ = G/n = G .
dG = - S dT + Vdp, при постоянной температуре dG T = Vdp.
Проинтегрируем последнее выражение от некоторого стандартного состояния до заданного с учетом уравнения Менделеева-Клапейрона
V = R T/p.
G |
p |
dp |
|
|
p |
|
|
|
∫dG = R T ∫ |
= R T ln |
|
|
|||||
p |
0 |
|||||||
G 0 |
p0 |
|
|
p |
||||
GT= G0T + RT ln p/p0. |
µT = µ0T + RT ln~p |
Обе записи эквивалентны. |
||||||
|
|
|
|
|
|
|
||
Так как p0 = 1 атм, то его часто опускают, но надо помнить, что под логарифмом не может стоять размерная величина, а только относительное давление ( или приведенное к стандартному давлению). При постоянной температуре
химический потенциал пропорционален логарифму приведенного давления, а
µ0T - это стандартный химический потенциал идеального газа при заданной
температуре ( и давлении = 1 атм)
8.5.2. Смесь газов Рассмотрим процесс смешения: исходное состояние – 2 газа в стандартных
условиях, конечное состояние смесь газов при той же температуре и том же
|
|
|
|
|
общем давлении. |
|
|||||
|
p0A, T, nA |
p0B, T, nB |
|
||||||||
|
|
|
|
|
|
|
|||||
|
PA ++ pB = p0, T, nA,nB |
|
|
|
|
||||||
|
p |
p |
RT |
|
|
|
|
pA |
0 |
|
|
∆G A = ∫ Vdp = n ∫ |
p |
dp = nRT ln |
|
; |
∆GB = nBRT ln pB/pB |
. |
|||||
0 |
|||||||||||
|
p0 |
p0 |
|
|
|
|
pA |
|
|
||
∆G = ∆G A + ∆GB |
= RT(nA ln pA/pA0 + nB ln pB/pB0) |
|
|||||||||
∆GpT = nA(µA - µA |
0) + nB |
(µA - µA0). Отсюда |
|
||||||||
µA = µA0 + RT ln pA/pA |
0 и µB = µB0 + RT ln pB/pB0, т.е. химический потенциал |
||||||||||
компонента идеальной газовой смеси зависит от парциального давления этого
компонента µk = µk0 + RT ln ~pk/ 8.5.3. Реальные газы
8.5.3.1. Уравнение состояния реальных газов. Уравнение Ван-дер-Вальса.
Уравнение В-д-В включает поправки на объем самих молекул ( силы
отталкивания) и взаимодействие молекул (притяжение).
http://www.mitht.org/forum/
27
Р |
|
|
|
|
|
|
|
|
( p + |
|
a |
) (V – b) = R T |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
V 2 |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Уравнение кубическое – оно имеет 3 |
||||
|
|
|
|
|
|
|
|
|
корня. Для изотермы Т 1 - L, O, G . По |
||||
|
|
|
|
|
|
K |
|
мере повышения температуры эти точки |
|||||
|
|
|
|
|
|
|
|
|
сближаются, а при температуре Т к слива- |
||||
|
|
|
|
|
|
|
|
|
ются в одну точку К, называемую крити- |
||||
|
|
|
|
|
|
N |
|
ческой, в ней все корни вырождены - они. |
|||||
|
|
|
L |
G |
Т к |
имеют одно значение V 1 = V 2 = V 3 = V крит. |
|||||||
|
|
|
|
|
|
O |
|
При дальнейшем повышении температуры |
|||||
|
|
|
M |
|
|
T 1 |
два корня становятся мнимыми. При |
||||||
|
|
|
|
|
|
|
|
|
T> |
T крит. газ нельзя перевести в жидкость |
|||
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
A D |
E B |
V |
изотермическим сжатием. При T< T крит , |
|||||||
напротив, |
газ переходит в жидкость при сжатии, и область под колоколом АКВ – |
||||||||||||
это область двухф азная – жидкость и пар находятся в равновесии. |
|||||||||||||
Так, при сжатии по изотерме Т 1 в |
точке G начинается конденсация пара, и |
||||||||||||
дальнейшее сжетие в реальности не приводит не к увеличению давления, а к увеличению количества жидкости – давление насыщенного пара постоянно. Таким образом реальный процесс конденсации пара изображается прямой линией G L, а не кривой ван-дер-Ваальса. Линия G N отвечает метастабильным неравновесным состояниям переохлажденного пара, а участок LM - перегретая жидкость. Область DMKNE – нераелизуемые состояния, когда в увеличением давления мольный объем газа увеличивается.
Критические параметры связаны с коэф ф ициентами a и b уравнения в-д-
В, которые имеют индивидуальные значения для каждого газа: V крит. = 3b ; T крит. = |
|
8а/27 bR ; |
pкрит. = a/27b2. Поэтому если мы будем вместо обычных |
термодинамических параметров использовать их приведенные значения, т.е. отнесенные к критическим, томожно для всех газов написать одно более общее
уравнение (π + 3/ϕ2) (3ϕ-1) = 8 τ, где π = р/ркрит. ; ϕ = V/V крит. ; τ=Т/Т крит. Э то уравнение справедливо для различных веществ, имеющих сходный характер
взаимодействия молекул. Оно показывает, что при одинаковых приведенных параметрах газы ведут себя сходным образом, и такие состояния называют соответственными.
8.5.3.2. Химические потенциалы реальных газов.
dG T = dµ = Vdp. Чтобы проинтегрировать это уравнение нужно было бы иметь аналитическую зависимость мольного объема от давления для реального газа, но кубическое уравнение не имеет общего решения, поэтому нельзя представить такой зависимости, поэтому Льюисом был предложен совсем другой подход, который состоит в следующем: для реальных газов остается такая же ф орма термодинамических соотношений, что и для идеальныхбгазов, но вместо давления используется другая переменная, называемая леутчестью, которая позволяет правильно описать свойства реальных газов
http://www.mitht.org/forum/

25
Лекция № 9
8.5.3.Р еальные газы
8.5.3.1.Уравнение состояния реальных газов. Уравнение Ван-дер-Вальса. Уравнение В -д-В включает поправки на объем самих молекул ( силы отталкивания) и взаимодействие молекул (притяжение).
Р |
|
|
|
|
|
|
|
( p + |
a |
) (V – b) = R T |
||
|
|
|
|
|||||||||
|
|
|
V 2 |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Уравнение кубическое – оно имеет 3 |
|||
|
|
|
|
|
|
|
|
|
корня. Для изотермы Т 1 - L, O, G . По |
|||
|
|
|
|
|
|
K |
|
мере повышения температуры эти точки |
||||
|
|
|
|
|
|
|
|
|
сближаются, а при температуре Т к слива- |
|||
|
|
|
|
|
|
|
|
|
ются в одну точку К, называемую крити- |
|||
|
|
|
|
|
|
N |
|
ческой, в ней все корни вырождены - они. |
||||
|
|
|
L |
G |
Т к |
имеют одно значение V 1 = V 2 = V 3 = V крит. |
||||||
|
|
|
|
|
|
O |
|
При дальнейшем повышении температуры |
||||
|
|
|
M |
|
|
T 1 |
два корня становятся мнимыми. При |
|||||
|
|
|
|
|
|
|
|
|
T> |
T крит. газ нельзя перевести в жидкость |
||
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
A D |
E B |
V |
изотермическим сжатием. При T< T крит , |
||||||
|
|
|
напротив, |
газ переходит в жидкость при сжатии, и область под колоколом АКВ – |
||||||||
это область двухф азная – жидкость и пар находятся в равновесии. |
||||||||||||
Так, при сжатии по изотерме Т 1 |
в точке G начинается конденсация пара, и |
|||||||||||
дальнейшее сжетие в реальности не приводит не к увеличению давления, а к увеличению количества жидкости – давление насыщенного пара постоянно. Таким образом реальный процесс конденсации пара изображается прямой линией G L, а не кривой ван-дер-Ваальса. Линия G N отвечает метастабильным неравновесным состояниям переохлажденного пара, а участок LM - перегретая жидкость. Область DMKNE – нераелизуемые состояния, когда в увеличением давления мольный объем газа увеличивается.
В критической точке изотерма имеет перегиб, т.е. первая и вторая производные от давления по объему равны 0. Э ти уравнения дают связь между
коэф ф ициентами a |
и |
b уравнения в-д-В, которые |
имеют |
индивидуальные |
|||
значения для каждого газа и критическими параметрами: V крит. = 3b |
; |
T крит. = |
|||||
8а/27 bR ; |
pкрит. |
= |
a/27b2. Поэтому если мы |
будем |
вместо |
|
обычных |
термодинамических параметров использовать их приведенные значения, т.е. отнесенные к критическим, томожно для всех газов написать одно более общее
уравнение (π + 3/ϕ2) (3ϕ-1) = 8 τ, где π = р/ркрит. ; ϕ = V/V крит. ; τ=Т/Т крит . . Э то уравнение справедливо для различных веществ, имеющих сходный характер
взаимодействия молекул. При критических параметрах все газы ведут себя сходным образом. Э то справедливо и в точках равноотстоящих от критических, т.е. при одинаковых приведенных параметрах газы проявляют одни и те же свойства, такие состояния называют с о о т в е т с т в е н н ы м и.
8.5.3.2. Химические потенциалы реальных газов.
dG T = dµ = Vdp. Чтобы проинтегрировать это уравнение нужно было бы иметь аналитическую зависимость мольного объема от давления для реального газа, но кубическое уравнение не имеет общего решения, поэтому нельзя представить такой зависимости, поэтому Льюисом был предложен совсем другой подход, который состоит в следующем: для реальных газов остается такая же ф орма термодинамических соотношений, что и для идеальных газов, но вместо давления используется другая переменная, называемая леутчестью, которая позволяет правильно описать свойства реальных газов.
Идеальные газы Р еальные газы
http://www.mitht.org/forum/

26
∆G = nR T ln p2 /p1 |
∆G = nR T ln f2 /f1 |
G = G 0 + R T ln p |
G = G 0 + R T ln f |
µ = µ0 + R T ln p |
µ = µ0 + R T ln f |
8.5.3.3.Коэф ф ициент летучести. С тандартные состояния.
µ |
|
|
|
Химический потенциал идеального гпза линейно |
|
|
|
||
|
|
|
|
зависит от логариф ма давления. Для реального |
µ0 |
|
|
газа зависимость будет криволинейной. |
|
|
|
|
С тандартное состояние идеального газа при |
|
|
|
|
||
|
|
|
|
давлении 1 атм, для реального газа – при |
0 |
lnf =0 |
ln p летучести , равной единице. На самом деле |
||
стандартное состояние одно и то же для обоих газов: f0 = p0 = 1. Для того, чтобы придти к стандартному состоянию реальный газ нужно расширить поизотерме до бесконечно малого давления, а затем сжать до р = 1 по изотерме идеального
газа. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
Чтобы определить летучесть, нужно давление умножить на коэф ф ициент |
||||||||||||||||||||
летучести: |
f = γp, или |
|
γ = f/p. |
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
С войства реальных и идеальных газов сближаются по мере уменьшения |
||||||||||||||||||||
давления |
|
limf/p p→0 = 1. γ p→0 = 1. Таким образом именно коэф ф ициент |
|
|||||||||||||||||||
активность выражает отклонение реального газа от идеального и связан с |
|
|||||||||||||||||||||
взаимодействиями, существующими в реальном газе. |
|
|
|
|
|
|||||||||||||||||
|
|
Отношение летучести к стандарнному состоянию называется активностью |
||||||||||||||||||||
a = f/f0 = p/p0. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
8.3.5.4. Методы определения летучести и коэф ф ициента летучести. |
|
|||||||||||||||||||||
a) dµT = dG = R T d(lnf) = V dp = (R T/p - α) dp, |
|
|
|
|
|
|
|
|
||||||||||||||
где α = V ид . –V реал. |
|
V реал. = |
V |
ид . - α |
|
|
|
|
|
|
|
|
||||||||||
2 |
|
2 |
|
|
|
|
2 |
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
∫dµ = ∫ R T dp − |
|
∫αdp |
= R T ∫d ln f |
|
|
|
|
|
|
|
|
|
|
|||||||||
1 |
|
1 |
p |
1 |
|
1 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
f1 → p1→ 0 |
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
RT lnp2 - RT lnp1 - ∫αdp = RT lnf2 – RT lnp1 = RT lnp2 + RT lnγ -RT ln p1. |
|
|||||||||||||||||||||
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p |
|
|
|
|
|
p |
|
|
|
|
|
|
|
|
|
|
|||
RT lnγ = - ∫αdp . |
|
ln γ = - 1/RT ∫αdp . |
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
0 |
|
|
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|||
α |
|
|
|
|
|
|
|
|
|
|
γ |
|
|
|
1.5 τ |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0.8 τ |
|
|
|
|
|
|
b) Метод соответственных состояний |
|
0.5 τ |
|
|
p |
|
||||||||||||||||
|
|
|
π |
|
|
|
|
|||||||||||||||
Разные газы ведут себя одинаково при одинаковых приведенных параметрах |
||||||||||||||||||||||
π , τ. Есть справочные номограммы |
|
|
|
|
|
|
|
|
|
|
||||||||||||
Таблица летучести и коэффициентов летучести азота при 0 С |
|
|
|
|
||||||||||||||||||
р, атм |
|
1 |
|
|
|
10 |
|
50 |
|
|
100 |
|
200 |
|
400 |
600 |
|
1000 |
||||
f |
|
0.9995 |
|
9.956 |
|
49.06 |
|
|
97.03 |
|
194.4 |
|
424.8 |
743.4 |
|
1.839 |
||||||
γ |
|
0.99955 |
|
0.9956 |
|
0.981 |
|
|
0.970 |
|
0.972 |
|
1.062 |
1.269 |
|
1.839 |
||||||
9.Химические реакции и химическое равновесие
9.1Общее условие химического равновесия в закрытой системе
Вхимической реакции происходит изменение числа молей компонентов в
соответствии со стехиометрическим уравнением 0 =Σ νkAk. Этим системы с
химическими реакциями напоминают открытые системы, которые обмениваются
http://www.mitht.org/forum/

27
веществом с окружающей средой, поэтому полные дифференциалы
характеристических ф ункций следует записывать как для открытых систем, например, dG = - S dT + Vdp + Σµkdnk .
При p,T = const dG = Σµk dnk. Так же будут выглядеть диф ф еренциалы других характеристических ф ункций при постоянных значениях их естественных параметров. В самопроизвольных процессах при p,T = const энергия Гиббса должна убывать dG р,Т < 0, , а в состоянии равновесия ее изменение должно равняться 0
dG р,Т = 0, следовательно в самопроизвольном процессе при p,T = const
Σµk dnk < 0. Но если в истинно открытой системе изменения чисел молей веществ независимы друг от друга, то в закрытой системе с химической реакцией они связаны между собой и могут быть все выражены через химическую переменную
dnk/νk = dξ и dnk = νk dξ nk = nk0 + νk ξ
dG pT = Σµk νk dξ . В самопроизвольном процессе при p,T = const производная по степени протекания химической реакции должна убывать, а в состоянии химического равновесия равняться 0
|
∂G |
|
= |
∑ |
µ |
|
ν |
|
< 0 |
самопроизвольный процесс |
|
|
|
k |
k |
||||||
|
|
|
|
|
|
|
|
|||
|
∂ξ pT |
|
|
|
|
|
|
|
|
|
|
∂G |
|
= |
∑ |
µ |
|
ν |
|
= 0 |
|
|
|
|
k |
k |
|
|||||
|
∂ξ |
|
|
|
|
|
|
|||
|
pT |
|
|
|
|
|
|
|
|
|
Общее условие химического равновесия Σµk νk = 0
Э то условие применимо для любых параметрах, выражая химический потенциал через любую характеристическую ф ункцию, но наиболее просто это можно сделать через энергию Гиббса.
∆G pT = Σµk νk = Σ G k νk = Σ G j νj - Σ νi G i = 0.
∆G pT = Σ∆f G j νj - Σ νi ∆f G i = 0.
9.2. Уравнение изотермы химической реакции 9.2.1. Гомогенные газовые реакции
Уравнение изотермы химической реакции показывает изменение энергии Гиббса за счет полного превращения реагентов в продукты в мольных количествах,
соответствующих стехиометрическим коэф ф ициентам при |
|
p,T = const и постоянном составе системы |
|
∆G pT = Σµk νk = Σ G k νk |
это мера химической неравновесности системы |
-∆G pT сродство, способность системы к химическому превращению |
|
Подставим в это уравнение выражения для химического потенциала |
|
µ = µ0 + R T ln p |
µ = µ0 + R T ln f |
∆G pT = Σ(µ0 k + R T ln pk) νk = Σµ0νk + R T lnΠ pkνk = |
|
∆G pT = ∆G T 0+ R T lnΠ pkνk = ∆G T 0+ R T ln κp. = ∆G T 0 + R T ln |
∏pνj |
Для |
||||||
j |
|
|
|
|
|
. |
||
|
|
|
|
|
реальных |
|||
∏p |
|
ν |
i |
|
|
|||
|
|
|||||||
|
|
|
|
|
|
газов вместо |
||
|
i |
|
|
|
|
|
|
давления в |
уравнении изотермы будет стоять летучесть |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
http://www.mitht.org/forum/
|
|
|
|
|
|
|
|
|
|
|
28 |
|
|
|
|
|
|
|
|
|
Под |
||||
|
∏f νj |
|
|
|
|
|
|
|||||
∆G pT = ∆G T 0 + R T ln |
j |
|
|
|
|
|
= ∆G T |
0+ R T lnΠ fkνk = ∆G T |
0+ R T ln κf.. |
|
знаком |
|
|
|
|
|
|
|
логариф ма |
||||||
∏f |
|
ν |
i |
|
|
|||||||
|
|
|||||||||||
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
стоят |
|||
|
i |
|
|
|
|
|
|
|
|
|
произвольные |
|
парциальные давления, которые можно изменять как угодно, но которые |
||||||||||||
|
||||||||||||
поддерживаются постоянными в ходе эксперимента. Э того можно достичь используя очень большую систему, состав который не изменяется в ходе химической реакции, или проточную систему с массообменом. Э ти давления должны быть выражены в атмосф ерах, т.е. приведены к стандартному значению, чтобы под логариф мом стояла безразмерная величина.
9.2.2. Гетерогенные реакции Такие реакции протекают на границе раздела ф аз. Р ассмотрим реакцию FeO + H 2 = Fe + H 2O
ν -1 -1 1 |
1 |
∆G pT = µFe + |
µ H2O - µ FeO - µH2 = µ0 Fe + µ0 H2O - µ0 FeO - µ0 H2 – R T ln pH2 |
µFe = µ0 Fe µтв = µ0 тв.
Конденсированные ф азы находятся в стандартном состоянии, поэтому под логариф мом в уравнении изотермы стоят давления только газообразных веществ. Для обычной температуры – только давление водорода, при высокой
температуре |
|
- отношение парциальных давлений воды и водорода. |
|||
9.2.3. Влияние давления на направление реакции |
|||||
Уравнение изотермы химической реакции позволяет определить, как изменяется |
|||||
направление реакции, если мы изменяемобщее давление в системе |
|||||
3/2 H 2 + 1/2N2 |
= NH 3 |
T = 500 K ∆G 0 500 = 9.26 кДж.моль. |
|||
С тандартное изменение энергии Гиббса положительноБ т.е. при стандартных |
|||||
условиях реакция не идет в прямом направлении. Е сли сильно повысить |
|||||
давление до 600 атм, т.е. парциальное давление каждого газа будет cоставлять |
|||||
200 атм, то знак ∆G изменится |
|||||
∆G = ∆G 0 + R T ln |
|
200 |
|
= 9260 - 8.314*500 ln 5.29 = -12600 Дж/моль. |
|
2003 / 2 2001 / 2 |
|||||
|
|
||||
С оотношение между веществами осталось то же, но увеличение давления сместило реакцию в соответствии с принципом Ле-Шателье: число молей газообразных веществ уменьшается ∆νгаз = -1.
http://www.mitht.org/forum/
31
Лекция № 10
9.3.Закон действующих масс 9.3.1. Константа равновесия
∏pνj
∆G pT = ∆G T 0+ R T lnΠ p*kνk = ∆G T 0 + R T ln ∏j p νi = 0.
i
В состоянии равновесия при p,T = const ∆G pT = 0
- ∆G T 0 = R T lnΠ p*k νk = f (T) Отношение равновесных парциальных давлений продуктов к давлению реагентов в степенях, соответствующих модулю стехиометрических коэф ф ициентов, является величиной постоянной при Т=const.
∏pк* νj
Kp = ∏j pк* νi = Π p*kνk = f(T)
i
Для гетерогенных реакций в константу равновесия входят только равновесные парциальные давления газообразных веществ. Константа равновесия величина безразмерная, так как давления должны быть приведены к выбранному стандартному давлению.
- ∆rGT0 = RT ln Kp
∆rG Tp = R T ln κ/K p - уравнение изотермы химической реакции, где κ - отношение произвольных парциальных давлений продуктов и реагентов.
9.3.2. С пособы выражения константы равновесия 9.3.2.1. Идеальный газ при постоянном давлении и температуре
∏pк* νj |
∏x k* νj |
||||
Kp = ∏pк* νi = Π p*kνk = f(T) |
= ∏x k* νi p∆νк общ. = Kxpобщ∆νк. |
||||
j |
j |
||||
|
|
|
|
|
|
i |
i |
||||
р*к = робщ.Nk = робщxk. Кх = f (T,pобщ.). |
|
|
|
||
Если ∆ν> 0, при р↑ Кх↓ ; если ∆ν< 0, при р↑ Кх↑. |
|
|
|
||
9.3.2.2. Реальный газ |
|
|
|
||
∏f к* νj |
|
|
|
||
Kf = ∏j f к* νi = Π f*kνk = f(T) = Kp Π γkνk. Kp = f(p,T)
i
9.3.2.3. Идеальный газ при постоянном объеме и температуре pV = nRT, p = nRT/V = cRT
∏Ск* νj (RT )Κνj |
= Π C*kνk (RT)Σνk= f(T) = Кс(RT)Σνk |
||
Kp = ∏Ск* νi (RT )Κνi |
|||
j |
|
||
|
|
|
|
i |
|
||
Σνk включает только газообразные вещества.
Константу Кс удобно использовать для газов, если реакцию проводят при постоянном объеме, а также для жидких растворов.
9.4. Влияние температуры на химическое равновесие
http://www.mitht.org/forum/
32
9.4.1. Вывод уравнения Вант-Гоф ф а
|
|
Для вывода потребуются 2 уравнения: |
|||||||
|
0 |
|
0 |
|
∂∆ |
r |
G 0 |
|
|
∆r G |
= ∆r H |
|
|
|
|
уравнение ГиббсаГельмгольца и |
|||
|
|
+ T |
∂T |
|
|||||
|
|
|
|
|
p |
|
|||
- ∆rG T 0 = R T ln K p уравнение изотермы химической реакции. |
||||
0 |
∂(− R T ln K |
p ) |
||
|
|
|
|
|
- R T ln K p = ∆r H + T |
∂T |
|
. |
|
|
|
|
p |
|
|
|
|
|
|
0 |
|
|
|
|
2 |
∂ ln |
K |
p |
|
||||
- |
R T ln K p = ∆rH |
- R T ln K p – R T |
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
∂T |
|
|
. |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p |
|
||
|
∂ ln K p |
|
∆r H 0 |
∂ ln K C |
|
|
∆r U 0 |
|||||||||||
|
|
|
= |
|
|
|
|
|
|
|
|
|
|
= |
|
|
|
|
|
∂T |
|
|
R T |
2 |
∂T |
|
|
|
|
R T |
2 |
||||||
|
p |
|
|
|
|
|
V |
|
|
|
|
|||||||
Э ти уравнения называют изобарой и изохорой Вант-Гоф ф а. 9.4.2. Анализ уравнения Вант-Гоф ф а
R T2 > 0. Изменение константы равновесия с ростом температуры определяется стандартной теплотой реакции.
Е сли ∆rH 0 > 0 реакция эндотермическая, то при T↑ K p↑; Е сли ∆rH 0 < 0 реакция экзотермическая, то при T↑ K p ↓. Такие изменения соответствуют принципу Ле-Шателье
9.4.3.Использование уравнения Вант-Гоф ф а
9.4.3.1.Определение ∆rH 0 по температурной зависимости константы равновесия
2 |
T 2 |
∆r H |
0 |
|
а) две точки в небольшом интервале температур, ∆rH 0= сonst ∫d ln K p |
= ∫ |
|
dT |
|
R T |
2 |
|||
1 |
T1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆r H 0 |
T 2 |
dT |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
= |
|
|
|
|
|
|
∫ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
T |
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
T1 |
|
|
|
|
0 (T |
|
|
|
) |
|
|
|
|
K |
2 |
|
∆ |
r |
H 0 |
1 |
|
|
1 |
|
|
|
|
∆ |
r |
H |
2 |
− T |
1 |
|
|||||
ln |
|
|
= |
|
|
|
|
|
− |
|
|
|
= |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
K 1 |
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
T 1 T 2 |
|
|
|
|
|
|||||
|
|
|
|
T 1 |
|
|
T 2 |
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
∆rH 0 = R |
|
ln K 2 / K 1T1T 2 . |
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
T 2 − T1 |
|
|
|
|
|
|
|
|||||||
b) много точек в небольшом интервале температур |
|
||||||||||||||||||||||||||
|
|
|
|
|
ln K p = - ∆rH 0/R T + const |
|
|
|
|
|
|
||||||||||||||||
ln K p |
∆rH 0 < 0 |
|
|
|
|
tg α = - ∆rH 0 / R |
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
В большом интервале температур зависимости могут быть кривыми ,
∆rH 0 > 0 тогда нужно брать тангенс наклона касательной.
http://www.mitht.org/forum/
33
1/T
9.4.3.2. Определение константы равновесия при другой температуре Известна константа равновесия при 298 К и ∆rH 0
a) малый интервал темпратур ∆rH 0 = const
|
∆ |
r |
H 0 |
|
1 |
|
|
1 |
|
|
|
∆r H 0 (T − 298) |
|
|
+ |
|
|
|
|
|
− |
|
|
= ln K 298 |
+ |
|
|
. |
|
|
R |
298 |
T |
298T |
||||||||||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b) большой интервал температур ∆rH 0 = f (T)
∫d ln K p |
= ∫ |
( ∆r H 0 |
0 + ∆aT + ∆b / 2 * T 2 |
+ ∆c / 3* T 3 − ∆c' / T ) dT |
. |
|
R T |
2 |
lnK p = - ∆rH 0 0 /R T + ∆a/R * ln T + ∆b/2R * T + ∆c/6R *T2 + ∆c’/2R T2 + C .
Зная конствнту равновесия при какой-либо температуре, можно найти постоянную интегрирования, а затем рвссчитывать константу при любой температуре.
9.5. Расчетные и экспериментальные методы определения ∆rG 0 и констант равновесия
9.5.1.По справочным данным по изсенению стандартной энергии Гиббса для реакции образования веществ. Так как нельзя определить абсолютное значение внутренней энергии, этого нельзя сделать и для энергии Гиббса, поэтому за уровень отсчета принимают стандартные состояния простых веществ, для которых энергию Гиббса полагают равной 0. В справочниках приводятся данные по энергиям Гиббса, отсчитанным от этого уровня. Тогда
∆rG 0 = Σjνj∆fG 0 j - Σi ν i∆fG 0 i = Σkνk∆fG 0 k
9.5.2.Е сли нет данных по ∆fG 0, то можно использовать справочные данные по
стандартным теплотам образования и абсолютным энтропиям веществ
∆rG 0 = ∆rH0 – T ∆rS 0,
где ∆rH0 = Σjνj∆fH0 j - Σi ν i∆fH0 i = Σkνk∆fH0 k
и∆rS 0 = ΣjνjS 0 j - Σi ν iS 0 i = ΣkνkS 0 k.
Эти уравнения показывают, что самопроизвольному протеканию реакции способствует
ее экзотермичность и рост энтропии, однако обычно, если ∆rH0<0, то
∆rS 0<0 также, т.е. энергетический и энтропийный ф акторы действуют в разных направлениях, причем роль энтропийного ф актора возрастает с повышением
температуры
∆rH0<0 и ∆rS 0<0 если ∆rH0 > T∆rS 0, тогда ∆rG 0<0 ; ln K > 0; K > 1;∆rH0 < T∆rS 0, тогда ∆rG 0>0 ; ln K < 0; K < 1.
9.5.3. По измерению полезной работы
-∆rG 0 T,p = (W’)max, например, в элетрохимии можно измерить Э ДС гальванического
элемента в обратимом процессе
∆rG 0 T,p = -E 0 F z, где
E 0 – стандартная Э ДС элемента, F – число Фарадея, z – количество электронов, в уравнении окислительновосстановительной реакции.
9.5.4. Э кспериментальное определение констант равновесия.
- ∆rG T 0 = R T ln K p. Положительное значение ∆rG T 0 не означает, что реакция вообще не может идти, она не может протекать в стандартных условиях, когда реагенты и продукты присутствуют в системе под давлением 1 атм. Но в отсутствии продуктов в начале реакции, реакция конечно пойдет до состояния равновесия и задача экспериментатора найти равновесную степень превращения, или равновес-ный выход или состав равновесной смеси при заданных исходных условиях.
Каждая реакция требует своего подхода для определения этого равновесного состояния системы. Для газовых реакций это делается очень просто, если изменяется
http://www.mitht.org/forum/
34
число молей газообразных веществ, т.е. изменяется реакционный объем при постоянном давлении. Для этого нужно составить материальный баланс химической
реакции: nk = nk0 + νkξ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Е сли n i0 = 1, то ξ = α |
|
νk |
|
|
|
NO 2 |
|
|
= NO |
+ |
1/2O 2 |
|||||||||||||||
|
|
|
|
|
|
|
|
-1 |
|
|
1 |
|
|
|
|
|
|
1/2 |
|
|
|
|
|
|||
|
|
|
|
|
|
|
nk0 |
|
|
|
1 |
|
|
0 |
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
Σnk*= 1 + 1/2α* |
|
nk* |
|
|
1 - ξ* = |
|
|
α* |
|
|
|
1/2α* |
|
|
||||||||||
|
|
|
|
|
|
|
|
1 - α* |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
pk* = pобщ. Nk* = pобщ |
nk*/Σ nk* |
pk* p |
|
|
1 − α* |
|
|
p |
|
|
|
|
α* |
|
p |
|
|
1 / 2α* |
||||||||
1 |
|
* |
|
1 |
|
|
|
* |
1 |
* |
||||||||||||||||
|
|
|
|
|
|
|
|
|
+ 1 / 2α |
|
+ 1 / 2α |
|
+ 1 / 2α |
|||||||||||||
|
p* |
1 / 2 |
|
|
|
|
α* α*1/2 (1 + 1/2 α* ) |
|
|
|
|
|
|
|
|
|
|
α*3/2 |
|
|
||||||
K = |
NO p* O 2 |
|
= p |
1 / 2 |
|
|
= p |
1 / 2 |
|
|
|
|
. |
|||||||||||||
|
p* NO 2 |
|
(1 + α* / 2)(2 + α* )1 / 2 (1 − α* ) |
|
|
|
(1 − α* )(2 + α* )1 / 2 |
|||||||||||||||||||
|
V*/V 0 = n*/n0 |
= (1 + 1/2α*) ; |
(V*- V 0 )/V 0 |
= 1/2α*. |
|
|
|
|
|
|
|
|
||||||||||||||
Измеряя относительное увеличение объема, можно найти равновесную степень превращения и константу равновесия, и , наоборот, зная кнстанту, легко найти равновесный выход в любых условиях.
9.5.5. По известным константам равновесия некоторых реакции, можно найти константу равновесия для любой линейной комбинации этих реакций.
|
FeO + C = Fe + C O |
K = ? |
∆rG 10 |
||
1 |
|
|
FeO + C O = Fe + C O 2 |
K 1 |
|
|
|
||||
1/2 |
|
|
2 C + O 2 = 2 C O |
K 2 |
∆rG 2 0 |
-1 |
|
|
C O + 1/2 O 2 = C O 2 |
K 3 |
∆rG 3 0 |
|
|||||
∆rG 0 = ∆rG 1 0 + 1/2∆rG 2 0 - ∆rG 30 |
|
K 21/2 K 3 -1 |
|||
ln K = ln K 1 + ½1/2lnK 2 – ln K 3 . |
K = K 1 |
||||
9.5.6. С татистические расчеты констант равновесия Все термодинамические ф ункции выражаются через стсатистические суммы
веществ Q = Σi gi exp(-εi/kT)/ µk = G k = -R T ln Q/NA + U 0 .
Q/V= q - статистическая сумма единицы объема
|
∏q j |
νj |
||||
K c = |
j |
|
|
|
exp(-∆rU0 0 /R T) |
|
∏q i |
|
νi |
|
|
||
|
|
|||||
|
|
|
|
|
||
|
i |
|
|
|
|
|
http://www.mitht.org/forum/
35
Лекция № 11
10.Гетерогенные системы. Фазовые равновесия.
10.1.Основные понятия
10.1.1.Ф а з а – однородная часть системы, отделенная от других поверхно-стью раздела и характеризующаяся уравнением состояния f (p,T,nk)= 0.
Вотсутствеи равновесия внутри ф азы параметры могут изменяться не-
прерывно, а на границе – скачком. |
|
||
10.1.2. К о м п о н е н |
т |
– вещество, которое может быть выделено из систе-мы и |
Примечание [ЮАВ1]: |
существовать вне ее, |
т.е. это независимые вещества. |
||
Р асчет числа компонентов – из общего числс веществ нужно вычесть число |
|
||
уравнений, которые связывают их концентрации. |
|
||
Такими уравнения могут быть выражения для константы равновесия, если в сис- |
|
||
теме могут протекккать химические превращения. |
|
||
С аС О 3 = С аО + С О 2 |
Кр = рС О2 . |
|
|
Один и тот же набор веществ может иметь разное число компонентов
N 2 , H 2, NH 3 в отсутствии химического равновесия - 3 компонентная система, так как реакция в силу кинетических особенностей может протекать только при высокой температуре в присутствии катализатора.
При химическом равновесии система может вести себя как двухкомпонент-ная.
N 2 + 3 H 2, = 2 NH 3 , так как появится уравнение Кр = П рл νк.
Е сли же азот и водород появляются только за счет разложения аммиака
NH 3 = N 2 + 3 H 2 , то появляется еще одно уравнение, связывающее количества образующихся nN2 : nH2 = 1:3. Такие уравнения относятся к начальным условиям. В последнем случае система однокомпонентна.
Таким образом k = m – r – b, где m – число веществ ( видов частиц), К – число равновесных реакций, и – число уравнений связи.
10.1.3.С тепени свободы – число независимых переменных, которые можно изменять не меняя числа ф аз.
10.1.4.Классиф икация
|
Название по ϕ |
Название по k |
Название по v |
|
|
|
|
0 |
- |
|
Нон(ин)вариантная |
1 |
Гомогенная |
Однокомпонентная |
Моновариантная |
2 |
Двухф азная |
Бинарная, двухкомпонентная |
Бивариантная |
3 |
Трехф азная |
Тройная |
Тривариантная |
много |
Полиф азная |
Многокомпонентная |
Поливариантная |
10.1.5. Общие условия фазового равновесия Для гетерогенных систем в качестве определяющих параметров используются интен-
сивные параметры р и Т, объем не используется никогда, поскольку нужно было бы указывать объем каждой фазы, а они изменяются при фазовых переходах. Удобнее в качестве экстенсивных величин использовать числа молей компонентов.
T = const система находится в тепловом равновесии, p = cоnst - “ - механическое - “ -,
при расчете числа компонентов мы принимали во внимание химическое равновесие
Σνkµk = 0.
В гетерогенной системе есть еще один вид равновесия = фазовое. В отсутствие фазового равновесия происходит перенос вещества из одной фазы в другую, т.е. совершается химическая работа δWхим.. = - µkdnk - работа переноса вещества k. Изменение энергии Гиббса в фазе α
http://www.mitht.org/forum/
|
|
36 |
dG α= -S αdT + V αdp + µk αdnkα ; |
dG β= -S βdT + Vβ dp + µk |
βdnkβ . |
Общее изменение энергии Гиббса |
при р,Т = const dG pT = dG |
α + dG β |
В состоянии равновесия изменение энергии Гиббса равно 0. |
|
|
dG pT = µk αdnkα + µk βdnkβ = 0.
Так как происходит перенос вещества из ф азы α в ф азу β , то dnkα = - dnkβ и условием равновесия будет
µk α = µk β равенство химических потенциалов любого компонента во всех ф а- зах.
Таким образом за ф азовое равновесие ответственен одинаковый интенсивный ф актор - химический потенциал, также как в тепловом и механическом равновесии. В отсутствие ф азового равновесия перенос осуществляется из ф азы с более высоким химическим потенциалом в ф азу с более низким химическим потенциалом , что соответствует общему снижению энергии Гиббса dG = (µk2 - µk1) dnk.
10.1.6. Правило ф аз Гиббса определяет число степеней свободы гетерогенной системы. Для этого из общего числа переменных, описывающих систему нужно вычесть число уравнений, связывающих эти переменные.
Переменные |
- интенсивные – общие для всех ф аз |
р, Т |
2 |
|
|||||
|
|
Э кстенсивные – для каждой ф азы |
nkα |
kϕ |
|
||||
Общее число переменных |
|
|
|
|
kϕ + 2 |
||||
Уравнения |
- Уравнения состояния для каждой ф азы |
ϕ |
|||||||
Уравнения ф азового равновесия µk α = µk |
β |
|
ϕ - 1 |
||||||
Для каждого компонента независимых уравнений |
|
||||||||
Для всех компонентов |
|
|
|
|
k(ϕ - 1) |
||||
Степени свободы kϕ + 2 - ϕ - k(ϕ - 1) |
= kϕ + 2 - ϕ - kϕ + k = k + 2 - ϕ = v |
||||||||
|
|
|
v= k + 2 |
- ϕ |
|
правило ф аз Гиббса |
|||
Е сли система помещается в какое либо поле появляются дополнительные интенсивные характеристики, то вместо 2 будет ф игурировать 3. Е сли какой либо интенсивный параметр закрепляется, то 2 заменяется на 1.
10.1.7. Принципы ф изико-химического анализа 10.1.7.1. Задачи ф изико-химического анализа. С остояние гетерогенной системы
чаще всего представляется в виде диаграмм, Э то наиболее экономный и компактный способ представления, который позволяет понять, как будет вести себя гетерогенная система в тех или иных условиях. С уществует целый раздел термодинамики, называемый геометрической термодинамикой, в задачу которого входит представление состояний системы в виде наглядных геометрических образов, а с другой стороны применение методов математического аппарата и геометрии к анализу реальных взаимоотношений между ф ормами существования веществ, т.е. связь между ф изико-химическими свойствами системы и геометрическими ф ормами пространства. Н.С .Курнаков.
10.1.7.2. Принцип соответствия Каждой ф азе или набору ф аз соответствует определенный геометрический об-
раз – объем, поверхность, линия, точка, которые могут описываться уравнениями.
10.1.7.3. Принцип непрерывности
http://www.mitht.org/forum/

37
До тех пор, пока существует определенный набор ф аз, изменение свойств системы под действием внешних ф акторов происходит непрерывно . При изменении числа ф аз свойства системы изменяются скачкообразно.
10.2. Однокомпонентные системы 10.2.1. Применение правила ф аз к анализу однокомпонентных систем v= 1 + 2 - ϕ = 3 - ϕ.
Максимальное количество ф аз, находящихся в равновесии равно 3, максималь-
ное число степеней свободы |
|
= 2. Э то означает, чо все состояния однокомпонент- |
||
ной системы можно изобразить на плоскости. |
||||
Е сли ϕ = 1, то v = 2 – область на плоскости, |
||||
ϕ = 2, то v = 1 |
- линия |
- |
“ |
-, |
ϕ = 3, то v = 0 |
– точка |
- |
“ |
-. |
10.2.2. Диаграммы состояния однокомпонентных систем В качестве независимых переменных можно выбрать любые 2 параметра, лучше
интенсивные , например, р,Т; |
µ, Т, р,V |
µ |
Для каждой ф азы есть область |
|
|
|
|
|
∂µ |
= −S изменения параметров, при кото- |
|
Обл. |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
∂T p |
|
|
тв. |
Обл. |
|
|
|
|
рых данная ф аза устойчива. На- |
|
жидк. |
Обл. |
S тв. <S ж. < S пара |
|||||
Т пл. |
Т кип. |
пара |
|
|
|
пример, при низкой температуре |
|
|
|
|
|
устойчивой будет твердая ф аза, так как ее химический по- |
|||
|
|
Т |
|
||||
тенциал ниже, чем жидкости. При температуре плавления и кипения в равновесии находятся 2 ф азы, химические потенциалы их одинаковы, перенос вещества происходит при постоянной температуре при постоянном давлении.
Граф ики химических потенциалов ф аз, выходящие за пределы области существования ф азы отвечают неустойчивым метастабильным состояниям.
Более распространенной является диаграмма рТ
р |
|
Каждой ф азе соответствует область |
||
|
плавление |
К |
существования, двухф азные равнове- |
|
|
ж. |
|
сия описываются линиями, а три ф а- |
|
|
тв. |
|
сосуществуют в одной точке, которая |
|
|
испарение так и называется тройная точка О. |
|||
|
О |
|
Так как вещество может существовать |
|
|
Пар |
|
в разных кристаллических модиф и- |
|
|
сублимация |
|
|
кациях, на диаграмме может быть не- |
|
|
|
сколько тройных точек, но 4 ф азы со- |
|
|
|
|
|
существовать не могут. |
|
|
|
|
|
10.2.3.Уравнение Клапейрона-Клаузиуса описывает двухф азные равновесия в однокомпонентных системах.
10.2.3.1. Вывод
µ α = µβ ; µ = G; dµ =d G = - S dT + Vdp dµ α = dµ β в равновесии
- S α dT + Vαdp = - S βdT + Vβdp (Vα - Vβ) dp = ( S α - S β)dT
http://www.mitht.org/forum/

38
dp |
= |
(S α |
− S β ) |
∆S ф.п. |
= |
∆H ф.п. |
|
|||
|
|
|
− V β ) = |
|
|
|
|
Изменение давления при из- |
||
|
(V α |
|
||||||||
|
|
|
||||||||
dT |
∆ |
V ф.п. |
|
Тф.п. V ф.п. |
||||||
менении температуры = tgα наклона касательной к линиям двухф азных равновесий.
10.2.2.2. Применение уравнения Клапейрона-Клаузиуса к анализу диаграмм
жидкость |
|
|
|
|
|
|
|
пар ∆H испар. > 0; |
|
|
∆V = V пар – V ж >> 0; |
dp |
> 0 |
|||||||||||||||
|
|
|
|
|
|
|
|
dT |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
твердая ф аза |
|
|
|
|
|
|
пар |
∆H субл. |
> 0; |
|
∆V = V пар – V тв. >> 0; |
|
||||||||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||||||
твердая ф аза |
|
|
|
|
|
|
жидкость ∆H пл.. |
|
> 0; ∆V = V ж. – V тв. > 0 |
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
вода, висмут, таллий |
|
∆V = V ж. – V тв. < 0 |
dp |
< 0 |
||||||||||||||
220 атм |
|
|
|
|
|
|
|
|
|
|
|
K |
|
|
Можно рассчитать, какое |
dT |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
давление необходимо, чтобы температура |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
плавления понизилась на 1 о |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆H пл.. = 6,01 кДж/моль |
|
|
|||||||||
1 атм |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ρтв. = 0,9168 г/см3 V тв = 18/0,9168 = |
|||||||||||||
4,58 |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
=19,633 см3/моль |
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
мм Hg |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ρж. = 0,9998 г/см3 V ж = 18 см3/моль |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
0,006 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆V = -1,63 см3/моль |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
атм |
273,16 373,15 647.3 |
Т,К |
|
|
|
||||||||||||||||||||||
|
p |
T |
∆H |
|
|
|
|
|
|
|
|
|
∆H |
|
|
|
T |
|
|
|||||||||
|
∫dp = ∫ |
|
|
|
|
|
dT ; |
p –po |
= |
∆V |
|
ln |
|
|
|
; |
|
|
||||||||||
|
|
|
|
|
T |
o |
|
|
||||||||||||||||||||
p0 |
T o ∆V T |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
6010Дж / моль |
ln |
272 |
= 13520656Па = 133,5атм |
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
273 |
|
|
|||||||||||||||||
1,63* |
10−6 м3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
0. 2.2.4. Р авновесие жидкость |
|
|
|
|
|
|
пар |
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||||||
http://www.mitht.org/forum/
|
|
|
|
|
|
|
|
|
38 |
|
|
(S α − S β ) |
|
∆S ф.п. |
|
Лекция № 12 |
|||
dp |
= |
|
= |
∆H ф.п. |
|
||||
|
(V α − V β )= |
|
|
|
|
Изменение давления при изменении |
|||
|
|
|
|||||||
dT |
∆ |
V ф.п. |
|
Тф.п. V ф.п. |
|||||
температуры = tgα наклона касательной к линиям двухф азных равновесий. 11.3.2. Применение уравнения Клапейрона-Клаузиуса к анализу диаграмм
жидкость |
|
|
|
|
|
|
|
пар ∆H испар. |
> 0; |
|
|
|
∆V = V пар – V ж >> 0; |
dp > 0 |
|
|
|||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
dT |
|
|
|
|
|
твердая ф аза |
|
|
|
|
|
|
|
пар |
|
∆H субл. |
> 0; |
|
|
|
∆V = V пар – V тв. >> 0; |
|
|
|
|
|
|||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||
твердая ф аза |
|
|
|
|
|
|
|
жидкость |
∆H пл.. |
> 0; ∆V = V ж. – V тв. > 0 |
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
вода, висмут, таллий |
|
|
∆V = V ж. – V тв. < 0 |
dp |
< 0 |
|
|
|
||||||||||||||||||||||||||||
220 атм |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
K |
|
|
|
|
Можно рассчитать, какое |
|
dT |
|
|
|
|
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
давление необходимо, чтобы температура |
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
плавления понизилась на 1 о |
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆Hпл.. |
|
= 6,01 кДж/моль |
|
|
|
|
|
|
|
|
|||||||||||||||
1 атм |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ρтв. = |
0,9168 г/см3 V тв = 18/0,9168 = |
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
4,58 |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
=19,633 см3/моль |
|
|
|
|
|
|
|
|
||||||||||||||||||
мм Hg |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ρж. = 0,9998 г/см3 V ж = 18 см3/моль |
|
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
0,006 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆V = -1,63 см3/моль |
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
атм |
|
|
|
273,16 373,15 647.3 |
Т,К |
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||
|
p |
|
|
|
|
T ∆H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∆H |
|
|
|
T |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
∫dp = ∫ |
|
|
|
|
|
|
dT ; |
|
p –po |
|
= |
∆V |
|
ln |
|
|
|
|
|
; |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
T |
o |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
|
p0 |
|
|
|
|
T o ∆V T |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
6010Дж / |
моль |
ln |
272 |
|
= 13520656Па = 133,5атм |
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||
|
|
1,63* |
|
|
|
|
|
|
|
|
|
273 |
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||
|
|
10−6 м3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||
11.3.3. Р авновесие жидкость |
|
|
|
|
|
|
|
|
пар |
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||
11.3.3.1. Вывод уравнения Клаузиуса-Клапейрона |
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||
|
dpнасыщ. |
= |
|
∆H испар. |
= |
|
|
|
∆H испар. |
|
= |
|
∆H испар.p |
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||
|
|
dT |
|
|
|
|
∆V T кип . |
V парT кип . |
|
|
|
|
|
|
R T 2 |
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
∆V = V пар – V ж |
|
≈ V пар = R T/p. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||
|
d ln pнасыщ. |
|
= |
|
|
∆H испар. |
|
|
Уравнение КлаузиусаКлапейрона выражает зависи- |
||||||||||||||||||||||||||||||||||||||||||
|
|
dT |
|
|
|
|
|
|
R T 2 |
|
|||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
мость упругости ( давления насыщенного) пара оттемпературы и напоминает |
|||||||||||||||||||||||||||||||||||||||||||||||||||
уравнение Вант-Гоф ф а. Для равновесия жидкость – пар |
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||
Кр = рнасыщ, = р*. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
11.3.3.2. Интегрирование уравненеия Клаузиуса-Клапейрона |
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||
а) Определенный интеграл |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(T |
|
|
|
) |
|||||||||||||||||||||||||
|
2 dp |
|
|
|
2 ∆H |
|
|
|
|
|
|
|
|
|
|
|
|
p |
|
|
|
|
∆H |
|
1 |
|
|
1 |
∆H |
испар. |
2 |
− T |
1 |
||||||||||||||||||
|
|
|
= |
|
|
|
|
|
|
dT ln |
|
|
2 = |
|
|
|
|
|
|
|
|
|
|
|
− |
|
= |
|
|
|
|
|
|||||||||||||||||||
|
∫ |
|
∫ |
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
|
p |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
p1 |
|
|
R |
|
|
|
|
|
|
|
|
R T 1T 2 |
|
|
|
|
||||||||||||||||
1 |
|
|
|
1 R T |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
T1 |
T 2 |
|
|
|
|
|
||||||||||||||||||||||||
http://www.mitht.org/forum/
39
Это уравнение дает возможность
1)пересчитывать давление насыщенного пара с одной температуры на другую, если известна теплота испарения,
2)Определять теплоту испарения, если известна упругость пара при двух температурах;
3)Определять температуру кипения жидкости при разных давлениях Кипение - процесс образования пара внутри жидкости, в отличие от процесса
испарения, который происходит при любой температуре с поверхности жидкости.
Кипение возможно только тогда, когда давление насыщенного пара сравняется с внешним давлением. Условие кипения жидкости можно записать так р* = рнасыщ. =
рвнешн. |
|
|
|
|
p2 |
||
|
1 |
= |
1 |
− |
R |
ln |
|
|
|
∆H |
|
||||
|
T 2 |
T1 |
|
|
p1 |
||
Неопределенный интеграл для определения ∆H испар ln p = − R∆HT + const
lnp
∆H испар. > 0 ; tgα = - ∆H испар/R
1/Т
11.3.4. Правило Трутона
∆H испар можно также оценить на основании метода соответственных состояний, а именно на том, что состояния кипения различных жидкостей при одинаковом давлении , например стандартном давлении ро = 1 атм, являются соответственными, так как имеют примерно одинаковые приведенные значения температуры Т
Т крит. = τкип. = const. |
Вещества ведут себя сходным образом и следовательно |
∆S испар. = ∆H испар/Т кип. |
= const ≈ |
≈ 85 - 90 Дж/моль К Э то положение носит название правила Трутона. Оно справедливо для неассоциированных жидкостей, без дополнительных взаимодейст-
вий, таких как водородные связи в воде. |
|
|
|||
Вещество |
С C l4 |
H 2S |
C 6 H 6 |
C 6 H 12 |
H 2 O |
∆S испар, Дж/моль К |
86 |
88 |
87 |
85 |
109 |
12. Двухкомпонентные системы. Р авновесие твердая ф аза-жидкость.
12.1. Правило ф аз для двухкомпонентных систем υ = k + 2 - ϕ = 2 + 2 - ϕ = 4 - ϕ
υmin = 0, ϕmax |
= 4. |
В равновесии могут находиться 4 ф азы (соль, лед, раствор, |
пар). |
|
|
ϕmin = 1; υ max |
= 3. |
Три независимых переменных требуют посторения объемной |
диаграммы состояния, поэтому используется метод сечений, т.е. закрепляют один из интенсивных параметров р или Т. В этом случае диаграммы удается сделать плоскими, а правило ф аз для них можно записать в виде
υ = k + 1- ϕ = 3 - ϕ при р = const
ϕ = 1; |
υ = 2 |
области на диаграмме |
ϕ = 2; |
υ = 1 |
линии на диагграмме |
http://www.mitht.org/forum/
40
ϕ = 3; υ = 0 точки на диаграмме С остав ф аз можно выражать по-разному, например, в мольных долях:
NA = nA /(nA + nB ); NB = nB /(nA + nB ); NA + NB = 1. NA = 1 – NB .
Используются также весовые %, для пересчета нужно учитывать молярную массу
компонента nк = весовые % к/ Мк. |
|
|
|
|
|
|
|||||||||||
12.2. |
|
Анализ диаграмм |
плавкости, |
где ф вердыми ф азами являются чистые |
|||||||||||||
вещества. |
|
|
|
|
|
С |
|
|
|
Т пл.B |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
||||||||||||||
Т |
|
D |
p = const |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
расплав |
|
|
|
|
|
|
|
|
|
|
|
|
|
T пл.А |
|
D’ |
υ = 2 |
|
С ’ |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
υ = 2 |
|
|
|
|
|
|
|||||
А’ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
А” |
|
D” |
|
F” |
|
|
|
|
|
|
|
|
|
|
|
|
|
S A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
D’” |
|
|
|
|
|
|
|
S B |
|
|
|
|||||
|
|
|
|
|
E |
|
|
|
|
|
|
|
|
|
|
|
|
NB =0 |
0.25 |
|
0.5 |
|
|
0.75 |
|
|
|
|
|
||||||
|
|
|
|
1.0 |
|
|
|
|
|||||||||
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
||||||||||
NA = 1 |
|
|
|
|
|
NB |
|
NA = 0 |
|||||||||
|
|
|
|
|
|
|
|
|
|||||||||
12.2.1.Общие принципы построения диаграмм двухкомпонентных систем Изображение состава ф аз или системы, по оси абсцисс в виде откладывания мольной доли одного из компонентов, количество другого определяется вычитанием из 1. По ограничивающим вертикальным линиям – чистые компоненты.
Вся диаграмма разбита на поля, каждому из которых соответствует ф аза или набор ф аз.
Каждая точка на диаграмме называется ф игуративной точкой - ее координаты соответствуют параметрам системы в целом – температура и общий состав, который остается постоянным при различных состояниях какого-либо образца.
12.2.2.Гомогенная область расплава лежит выше линии Т пл.А Е Т пл.В , называемой линией л и к в и д у с а. В одноф азной области система имеет 2 степени свободы, т.е. можно изменять и состав и температуру расплава. Каждая точка соответствует определенному состоянию гомогенной системы. Процесс, происходящий в системе изображается линией, например, охлаждение – линией DD’. Изменение температуры без изменения состава жидкости происходит непрерывно, пока существует 1 ф аза, в соответствии с принципом непрерывности.
12.2.3.Гетерогенная область. При достижении температуры, соответствующей пересечению линии охлаждения расплава с линией ликвидуса, начинается кри-
сталлизация, мы попадаем в двухф азную область T пл.А D’Е S A . Аналогичная картина будет, если начальный состав расплава лежит правее – при охлаждении смеси
С мы попадаем в двухф азную область Т пл.B С ’Е S B . В этих областях
υ = 1. Получается вроде бы логическое противоречие. Оно разрешается следующим образом: ф игуративные точки в гетерогенных областях не имеют ф азового смысла, т.е. не являются параметрами, описывающими состав равновесных ф аз. С о с т а в ы р а в н о в е с н ы х ф а з лежат на л и н и я х , о г р а н и ч и в а ю
щ и х |
д в у х х ф а з н у ю о б л а с т ь, в соответствии с правилом ф аз Гиббса. |
Точки, |
соответствующие параметрам равновесных ф аз, соединяются линиями, |
которые называются коннодами. На диаграммах двухкомпонентных систем конноды – горизонтальные линии, так как обе ф азы должны иметь одинаковую температуру. Для того, чтобы определить , какая твердая ф аза кристаллизуется из дан-
http://www.mitht.org/forum/
41
ного расплава, нужно провести горизонтальную линию через точку D’ на линии ликвидуса до противоположной границы области т. А’.Э той точке отвечает состав – 100% компонента А. Из расплава С выпадает чистый компонент В .
Процесс кристаллизации чистого компонента А из расплава D сопровождается изменением состава жидкой ф азы по линии D’E . Температура кристаллизации при этом будет непрерывно изменяться. Так, в точке D” ( линия D’D” выражает процесс охлаждения двухф азной системы в целом) мы имеем 2 равновесные ф азы – жидкую состава F” и твердую ф азу чистого компонента А.
12.2.4. Таким образом линия л и к в и д у с а, с одной стороны показывает зависимость температуры начала кристаллизации твердой ф азы от состава расплава, с другой стороны, - составы жидкой ф азы, находящейся в равновесии с твердой
фазой, в зввисимости от температуры. Поэтому она иназвывается ликвидусом.
12.2.5.Правило рычага. Фигуративная точка внутри двухф азной области делит конноду на отрезки, пропорциональные относительному количеству равновесных
фаз, составы которых лежат на противоположном конце конноды.
A”D |
количество жидкой ф азы |
= 0.06 = 0,2 |
||
D”F” = |
количество твердой ф азы |
|
0,29 |
|
При Т” в точке F” было бы 100% жидкой ф азыБ а в точке А” 100% твердой. 12.2.6. Линия солидуса. Э втектика.
Продолжим охлаждение смеси до линии T пл.А S A S B T пл.B , называемой линией с о л и д у с а. В точке D’” начинает выпадать в осадок вторая твердая ф аза, таким образом число ф аз становится равным 3, а число степеней свободы –0.
Состояния ф аз должны изображаться точками ( образ нонвариантной системы).
Э то точки S A и S B – составы твердых ф аз и точка Е состав жидкой ф азы. Все три точки лежат на горизонтальной линии, т.е. имеют одинаковую
температуру. Точка Е называется э в т е к т и к о й, она выражает состав жидкой ф азы, находящейся в равновесии не менее, чем с двумя твердыми ф азами.
Состав смеси, отвечающей эвтектике называется эвтектическим, он может быть при любой температуре, но эвтектика может существовать только при одной температуре – эвтектической. То же самое получается, если охлаждать смесь С . Температура системы, в которой находятся в равновесии 3 ф азы не может изменяться. Чтобы температура вновь начала изменяться, должна исчезнуть 1 ф аза – жидкая, останутся только две твердые ф азы, область существования которых лежит ниже линии солидуса.
http://www.mitht.org/forum/
42
Лекция № 13 Точка Е называется э в т е к т и к о й, она выражает параметры жидкой ф азы,
находящейся в равновесии не менее, чем с двумя твердыми ф азами. С остав смеси, отвечающей эвтектике называется эвтектическим, он может быть при любой температуре, но эвтектика может существовать только при одной температуре – эвтектической. То же самое получается, если охлаждать смесь С . Температура системы, в которой находятся в равновесии 3 ф азы не может изменяться. Чтобы температура вновь начала изменяться, должна исчезнуть 1
ф аза |
– жидкая, останутся только |
две твердые ф азы, область |
существования |
которых лежит ниже линии солидуса Т пл.А S A E S B Т пл.В . |
|
||
Е сли |
рассматривать в микроскоп |
шлиф ы твердых образцов, |
поученных при |
охлаждении смесей С и D, то окажется , что они имеют совсем разный вид. Из смеси D сначала впадает кристаллы компонента А, которые постепенно растут, а затем начинает кристаллизоваться эвтектическая смесь, в которой частицы веществ А и В перемешены на молекулярном уровне. В результате получабтся очень мелко кристаллическая структура.
|
Из смеси С сначала будут выпадать кристаллы компонента В другого вида, а затем |
||||||
мелкокристаллическая структура эвтектики. |
|
|
|
||||
Е сли вести процесс нагревания смеси из 2 ф аз, то картина будет обратная той, |
|||||||
которую мы рассмотрели : начало плавления лежит на линии солидуса, т.е. |
|||||||
выплавляется эвтектика, а заканчивается плавление на линии ликвидуса. Т.е. всегла |
|||||||
температура плавления чистого вещества выше, чем температура ближайшей |
|||||||
эвтектики, это дает основание для контроля за чистотой вещества. |
|||||||
|
12.3. Построение диаграмм плавкости по кривым охлаждениянагревания. |
||||||
Кривая охлаждения дает экспериментальную зависимость температуры от времени |
|||||||
при остывании образца. |
|
|
|
|
|
||
Т |
D |
C |
А |
D |
E |
C |
B |
|
|
|
υ =1 |
υ = 2 υ= 1 υ = 2 |
|||
Т плАυ |
|
υ = 0 |
υ =1 |
υ = 1 |
|||
|
|
|
|
|
|
||
|
|
|
|
|
υ =0 |
υ=0 |
|
|
|
E |
|
|
υ =0 |
|
|
|
|
|
|
|
υ =1 |
|
υ = 1 |
|
|
NB |
|
|
|
|
|
|
Когда |
υ = 0, на кривой охлаждения – остановка, площадка, когда переходбот υ = |
|||||
2 к υ = 1, на кривой перегиб, замедление охлаждения. |
|
||||||
12.4.Холодильные смеси – это смесь растворителя – воды - с солями, которые
используются для достижения низких температур< Т пл. чистого растворителя. Диаграмма в принципе такая же, как диаграмма плавкости, но линия ликвидуса не достигает Т пл. чистой соли, так как растворитель
http://www.mitht.org/forum/
43
может закипеть. Криогидратная точка – это параметрs ра-створа, находящегося в равновесии с 2 твердыми ф азами–льдом и со-лью. Для NaC l 22% cсоли, Т пл. = -21,2 С ; С aC l2 –55 C , ZnC l2 - 62 C
12.5.Диаграсммы плавкости в случае образования твердых растворов.
12.5.1.Типы твердых растворов. Р аствор – ф аза переменнго состава, состоящая как минимум из 2 компонентов. Твердые растворы замещения образуются, когда вещества имеют близкие
химические свойства и близкие параметры кристаллической решетки, близкие кристаллограф ические радиусы. Тогда в узлах кристаллической решетки вместо одних атомов могут становиться другие – хаотично размещаемые. Bi – S b, Ag – Au, K 2 S O 4 – R b2 S O 4
–могут смешиваться в любой пропорции в тв. Фазе.
Растворы внедрения. Частицывнедряемого вещества занимают междоузлия. Обычно размеры внедряемого компонента меньше, чем основного, В кристаллической решетке возникают напряжения, поэтому растворимость ограничена.
12.5.2.Диаграммы плавкости в случае полной смешиваемости в тв.ф азе
Т |
|
|
D |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
Ликвидус D’ |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
∆Т – интервал температур, в |
|||||
|
|
|
|
|
|
|
|
||||
|
|
|
D” |
|
|
|
|
котором |
происходит плав- |
||
|
|
|
|
|
|
|
|
|
|
||
|
|
С олидус |
|
|
|
|
ление. Э втектической |
|
|||
|
|
|
|
|
|
|
|
площадки нет. По мере |
|
||
|
|
|
|
|
|
|
|
выпадения тв. Р аствора β |
|||
|
|
|
|
|
|
|
|
состав жидкой ф азы |
|
||
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
NB |
изменяется по линии |
ликвидуса. Аg- |
||||
|
|
|
|
|
|||||||
Au ( 960-1063 o C ). |
|
|
|
|
|||||||
В некоторых случаях растворы определенных составоыв обладают |
|||||||||||
повышенной или пониженной устойчивостью, тогда диаграммы |
|||||||||||
разбивается на две простые (рыбки). В экстремальных точках линии |
|||||||||||
ликвидуса и солидуса соединяются. |
|
|
|
||||||||
Т |
|
p = const |
Т |
p = const |
|
||||||
C aS iO 3 |
MgS iO |
3 ZnC l2 |
P bC l2 |
12.5.3. Диаграммы плавкости в случае частичной смешиваемости образуются чаще всего растворами внедрения. Они бывают разных видов. Один
из них мы рассмотрим. Она представляет собой соединение 2 диаграмм 12.2. и |
|||||||
12.5.2. |
|
|
|
|
|
|
|
Т |
p = const |
|
|
|
Ag - C u |
||
υ = 2 |
υ = 1 |
||||||
|
Р асплав |
960 1083 |
|||||
|
|
|
|
|
|
E 779oC , 28.5 C u |
|
|
|
|
|
|
|
|
|
http://www.mitht.org/forum/

44
|
|
|
|
|
|
|
8,8 C u |
92 % C u |
|
|
|
|
|
|
υ = 1 |
||||
|
|
|
|
|
|
|
|
||
|
α |
β |
|
|
|
υ = 0 |
S n – P b, Bi-S n |
||
|
|
α + β |
|
υ = 1 |
KNO 3 - NaNO 3 NO 3 |
||||
|
|
|
|
|
|
|
|
|
|
P b |
|
Zn |
|
|
время |
|
|||
12.6.Диаграммы плавкости в случае образования химических соединений
12.6.1.Конгруэнтное плавление. Термин конгруэнтный означает совпадающий, т.е. состав расплава и раствора совпадают, соединение существует и в твердой, и в жидкой ф азах, хотя в расплаве может происходить диссоциация его на компоненты. Об образовании химического соединения свидетельствует максимум на кривой ликвидуса при наличии эвтектических площадок на кривых охлаждения. Э тот максимум тем острее, чем устойчивей химическое соединение. Точка максимума D называется дистектикой, в ней
число степеней свободы равно 0 (υ = 2 - 1 + 1 –2 = 0). Т p = const D
D
Распл. pасплав
Распл. +
+Zn |
|
|
|
|
|
|
расплав |
|
|
|
|
|
|
|
|||
E 1 |
. МgZn2 |
|
|
|
|
|
|
|
|||||||||
Zn +MgZn2 |
+Mg |
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E 2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
MgZn2 + Mg |
|
|
|
|
|
|
|
||||||
Zn |
|
|
|
|
|
|
|
Mg |
|
|
|
|
|
время |
|
||
12.6.2.Инконгруэнтное плавление наблюдается в случае образования |
|||||||||||||||||
неустойчивого химического соединения, которое распадается при |
|||||||||||||||||
температуре ниже, чем температура его плавления. |
|||||||||||||||||
|
Т |
|
|
p= const |
|
|
C |
|
|
|
D |
|
C |
||||
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
D |
|
|
|
|
|
|
|
||||
|
|
|
Жидкость |
|
Na |
жидкость |
|
|
|
||||||||
|
|
|
|
|
|
P |
|
тв. |
|
|
S |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
Жидкость Na |
тв |
|
|
|
|
|
|
|
|||
|
ж |
|
. |
|
+ Na 2 K тв. + Na 2 |
K тв |
|
|
|
||||||||
Тв. р-р |
|
α + αтв. |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
E |
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
αтв + Na 2 K тв. |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
K |
|
|
|
NNa |
|
|
|
Na |
|
время |
||||||
На линии Р S сосуществуют 3 ф азы: жидкость состава Р (перитектика), тв. химическое соединение Na 2 K и тв. Na. При охлаждении происходит перекристаллизация: исчезает тв. Na и появляется тв. Na 2 K .
http://www.mitht.org/forum/
45
Лекция № 14
13. Р астворы
13.1. Основные понятия 13.1.1. Виды растворов. Р аствор гомогенная система переменного состава, состо-ящая
более, чем из 2 компонентов. Компоненты часто называют растворителем (он присутствует в большем количестве) и растворенное вещество. Индекс растворителя –1, индекс растворенного вещества – 2.
Р астворы могут находиться в разном агрегатном состоянии: Газ – газ (газовые смеси), Твердое – твердое вещество ( твердые растворы),
Газ – твердое вещество ( водород в платине)
Жидкость – жидкость |
|
Жидкость - газ |
растворы на основе жидкой ф азы занимают |
Жидкость – твердое вещество центральное место.
Жидкость – это конденсированная ф аза с квазикристаллической структурой, плотной упаковкой и сильным межмолекулярным взаимодействием. С илы межмолекулярного взаимодействия могут быть ф изическими ( Ван-дер-Ваальса) и химическими (например, водородные связи)
13.1.2. С пособы выражения состава раствора
Мольная доля |
Nk = nk/ Σnk} |
Nk |
= xk в растворе |
|
Nk |
= yk |
в равновесном паре. |
Весовая доля |
mk/Σmk , если умножить на 100, получится весовой процент. |
||
Молярность |
М = nk/V раствора количество молей вещества в единице объема рас- |
||
|
|
|
твора. |
Моляльность |
m = nk/m1(кг) |
количество молей вещества в 1 кг растворителя. |
|
13.1.3. Парциальные мольные величины Парциальная мольная величина – это производная от любой экстенсивной ха-
рактеристики системы Z по числу молей данного компонента при постоянных p, T и составе раствора, она показывает изменение этой экстенсивной величины при введении в раствор 1 моля данного компонента при постоянных условиях, т.е в бесконечно
|
∂Z |
|
|
|
большую систему Z = |
|
, в теории растворов парциальные мольные величи- |
||
|
||||
|
|
|
|
|
|
∂n k p,T ,n l |
|
||
ны заменяют обычные мольные характеристики экстентивных переменных. В качестве Z могут выступать любая экстенсивная величина
Z = V,S ,U,H,A,G ,C . Понятие парциальных мольных величин вводится, чтобы сохранить ф орму уравнений аддитивности, которые обычно применяются для идеальных смесей, с которыми мы обычно имели дело до сих пор. Уравнения аддитивности означают, что
каждый компонент вносит вклад |
в общую экстенсивную хврактеристику |
пропорциолнально своему количеству: |
|
Z = Z10 n1 + Z2 0 n2 , где Z1 0 – мольная характеристика чистого вещества ( стан-дартное состояние – это состояние чистого вещества в наиболее устойчивой ф орме под давлением 1 атм, при этом газ облаюает свойствами идеального газа, а конденсированные ф азы находятся в расновесии со своим паром. Таким образом мы
расчитывали энергию Гиббса G = Σµknk = ΣG knk, Объем V = V A 0nA + V B 0nB .
Е сли экстенсивную величину поделить на общее число молей в системе, то получим среднюю мольную величину
http://www.mitht.org/forum/

|
|
|
|
46 |
Z/Σnk = ZA 0 NA + ZB 0 NB |
= Z |
ср. = ZA 0 (1 – NB ) + ZB 0 NB = ZA 0 + NB (ZB |
0 - ZA 0) –получили |
|
уравнение касательной: ZA 0 |
– отсекаемый отрезок на оси z, а ZB 0 - ZA |
0 |
||
Z |
ZB 0 |
тангенс наклона. На осяхz отсекаются пар |
||
p,T= const |
|
|
циальные мольные величины. Они зависят |
|
|
ZB |
|
от состава раствора и могут отклоняться от |
|
|
|
|
стандартных мольных величин как в ту, так |
|
ZA 0 |
|
|
и в другую сторону. |
|
|
|
По определению химический потенциал - |
||
|
|
|
парциальная мольная энергия Гиббса. |
|
ZA
NB 
В неидеальных смесях парциальные мольные величины заменяют обычные мольные характеристики, которые можно использовать только для идеальных смесей, но ф орма уравнений остается при этом неизменной.
13.2. Идеальные растворы 13.2.1. Модель идеального раствора. С илы взаимодействия между частицами не
изменяются при образовании раствора, т.е. они равны для для чистых компокомпонентов и смеси: F AA =F BB =F AB . Э то означает, что при образовании раствора не происходит изменений внутренней энергии и объеме ∆V = 0; ∆U = 0
∆H = 0; но ∆S ≠ 0. Изменение энтропии будет определяться так же, как при образовании газовой смеси
∆S = - R (nA lnNA + nB ln NB ).
Важно определить изменение энергии Гиббса при образовании раствора. В него вносит вклад только энтропийный член:
∆G = ∆H - T∆S = R T(nA lnxA + nB lnxB ) < 0, так как мольная доля всегда меньше 1. 13.2.2. Химический потенциал компонента идеального раствора. Изменение энергии Гиббса при образован ии раствора можно также выразить через изменение химических
потенциалов компонентов при переходе от чистого состояния к раствору:
∆G рТ = G раствора – G чист.веществ = Σnkµk - Σnkµ0 k = Σnk(µk - µ0 k).
Для двухкомпонентного раствора ∆G рТ = nА (µА - µ0 А )+ nВ (µВ - µ0 В ). При
сравнении двух выражений для ∆G рТ видно, что R Tnklnxk = nk(µk - µ0 k)
µk = µ0 k + R T lnxk |
Химический потенциал компонента идеального раствора зависит от |
|
мольной доли этого компонента в растворе, а за стандартное со- |
|
стояние принимается состояние чистого компонента. Химический потенциал компонента в растворе всегда ниже, чем в чистом состоянии.
13.2.3. Давление насыщенного пара над раствором. Закон Р ауля.
Р ассматривая однокомпонентные системы мы видели, что давление насыщенного пара зависит только от температуры системы. Для растворов влияние на упругость пара оказывают в соответствии с правилом ф аз Гиббса (υ = 2 + 2 – 2 = 2) уже 2 ф актора -
температура и состав раствора. Р ассмотрим влияние состава раствора на упругость пара при постоянной температуре. Запишем условие ф азового равновесия µkраствор =
µkпар.
µ0 k(жидк.) + R T lnxk = µ0 k(пар)+ R T lnрk.
dµkраствор = dµkпар R T dlnxk = R Td lnрk d(lnxk) = d(lnрk).
Проинтегрируем это уравнение от чистого состояния до определенного состава раствора
http://www.mitht.org/forum/
47
x |
p |
|
∫d ln x = ∫d ln p ln x = ln p/p0 |
pk = pk0 x |
|
|
||
1 |
p0 |
|
Э то выражение представляет собой закон Р ауля, который справедлив для идеальных растворов и читается так : при постоянной температуре парциальное давление насыщенного пара компонента над раствором равно упругости пара над чистым компонентом, умноженным на мольную долю этого компонента в растворе.
Можно преобразовать закон Р ауля к другому виду:
p1 /p1 0 = x1 1 - p1 /p10 = 1 - x1 (p10 - p1 )/ p10 = x2
Относительное понижение давления пара растворителя над раствором равно мольной доле растворенного вещества. Э то уравнение удобно использовать, если растворенное вещество является нелетучим, тогда упругость пара растворителя заменяется общим давлением насыщенного пара над раствором.
13.2.4. Граф ическое выражение закона Р ауля.
рТ = const
|
жидкость |
|
|
|
|
pB 0 pобщ. = pA + pB = pA 0 xA + pB 0xB = |
|||
|
|
|
|
|
|||||
p = pA + pB |
|
п |
|
|
= pA |
0(1 – хВ ) + pB 0xB = |
|||
|
|
|
|||||||
|
|
ж + |
|
pB = pB 0xB |
|
= pA |
0 + xB (pB 0 - pA 0). |
||
pA 0 |
|
|
|
|
Э то уравнение общего давления над |
||||
|
|
|
|
|
|
|
раствором в зависимость от его состава. |
||
|
|
pA = pA 0 xA |
|
|
pA 0 – отсекаемый отрезок на оси ординат. |
||||
|
|
Пар |
|
|
|
|
(pB 0 - pA 0) = tg α. |
||
|
|
|
|
|
|
|
Выше линии общего давления система |
||
A |
|
|
|
|
|
|
|
находится в жидком состоянии. |
|
|
|
N |
B |
B |
|
||||
|
|
|
|
|
|||||
Е сли понижать внешнее давление над раствором, процесс изобразится вертикальной линией, при пересечении ее с линией общего давления будет выполняться условия
кипения жидкости робщ. = рвнешн. , и раствор закипит. Таким образом линия общего давления выражает зависимость упругости пара над раствором в зависимости от его
состава, или, наоборот зависимость составов кипящей жидкости в зависимость от внешнего давления. Так или иначе равновесие жидкость пар при постоянной температуре зависит от 1 переменной (υ = 2+1-2=1).
13.2.5. С оотношение составов пара и жидкости в идеальных растворах. Линия пара.
По закону Дальтона рk = робщ. Nk = робщ yk.
yB = рB / робщ = pB 0xB /[ pA 0 + xB (pB 0 - pA 0)]. Уравнение смещенной гиперболы.
рB = робщ yB .
Найдем соотношение составов пара и жидкости:
yB / xB = pB 0/[ pA 0 + xB (pB 0 - pA 0)] = 1/[a + xB (1-a)] = 1/[a(1- xB ) + xB ],
где а = pA 0/ pB 0.
Е сли pB 0> pA 0, то есть упругость чистого компонента В больше, чем упругость А.
то а < 1 и знаменатель < 1, тогда yB > xB , это означает, что в паре содержится больше
легколетучего компонента по сравнению с жидкостью. Например, xB = 0,5, yB = 0,5 pB 0/0,5(pА 0+ pB 0) > 0,5.
Таким образом мы получили для идеальных растворов 1-ый закон Коновалова:
http://www.mitht.org/forum/
48
Пар по сравнению с жидкостью обогащен компонентом, добавление которого повышает упругость пара над жидкостью, или понижает температуру кипения раствора. Пар обогащен легколетучим компонентом.
На этом законе основано разделение смесей методом перегонки и ректиф икации. Для расчета нужно воспользоваться другой диаграммой Т-состав,
которая представляет другое сечение трехмерной диаграммы состояния бинарной смеси.
13.2.6. Диаграмма кипения идеального раствора.
Т |
|
|
p = const |
|
|
|||||
|
|
|
||||||||
Т0 кип.А |
|
|
Пар |
υ = 2 |
Возьмем раствор состава х и будем его на- |
|||||
|
|
С ” |
гревать, этот процесс изобразится верти |
|||||||
|
|
|
|
|
|
|
|
|
кальной линией,. Когда эта линия пересе- |
|
|
|
ж. |
|
|
|
|
|
чется с линией жидкости, раствор закипит. |
||
|
|
C |
|
|
|
|
|
|
С истема станет двухф азной и число степе- |
|
|
|
|
+ |
C ’ |
|
|
|
|||
|
|
|
|
пар |
|
|
ней свободы уменьшится до 1. Е сли мы |
|||
|
|
|
|
|
υ = |
|
1 |
произвольно выбрали состав жидкости, то |
||
|
|
|
|
|
С ’” |
|
|
температура его кипения и состав образую- |
||
|
|
|
|
|
υ |
|
|
Т0 кип.В щегося пара определяются автометиче- |
||
|
|
жидкость |
|
= 2 |
ски: состав пара лежит на другом конце |
|||||
|
|
x |
|
|
y |
|
|
конноды, в нем содержится больше компо- |
||
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
А |
|
|
|
NB |
|
B нента В . При отводе пара жидкость обогащается |
||||
компонентом А, ее состав будет изменяться по линии жидкости влево, при этом температура кипения будет повышаться до температуры С ”, выше этой температуры вся жидкость перейдет в пар. Е сли отвести небольшую порцию пара начального состава и его сконденсировать, то его полная конденсация произойдет при температуре С ’”.
http://www.mitht.org/forum/

52
Лекция № 16 13.4.3. Химические потенциалы компонентов раствора
kB
p |
T= const |
kA |
poB |
poA |
|
1. Идеальные растворы во всей области составов – система симметричная, т.е. химические потенциалы компонентов выражаются обднаково через их мольные доли
А |
В |
µА = µА о + R T ln xA |
µB = µB o + R T ln xB . |
Стандартное состояние компонентов – это состояние чистого вещества. 2. Р азбавленные растворы
Система несимметричная – химические потенциалы компонентов выражются поразному:
Р астворитель |
Р астворенное вещество |
µ1 = µ1о + R T ln x1 |
µ2= µ2 о + R T ln m2 (µ2= µ2 о + R T ln х2 ) |
Химический потенциал растворителя зависит от его мольной доли в растворе, так как растворитель подчиняется закону Р ауля, и стандартное состояние растворителя – чистое вещество, на диаграмме это точка poA . Но для растворенного вещества стандартное состояние уже нельзя считать тпросто читсым компонентом В, так как закон Р ауля для него не действует. С тандартное состояние растворенного вещества – это его состояние в растворе, с концентрацией , равной 1, но сохраняющей свойства разбавленного раствора, т.е. свойства разбавленного раствора мы должны проэсктраполировать до концентрации, равной 1. Э то будет точка kB . Так как эта точка найдена искусственно, нет необходимости выражать его концентрацию растворенного вещества через его мольную долю, что не очень удобно. Поэтому принимают, что химический потенциал растворенного вещества зависит от концентрации раствора, а точнее - от его моляльности. С тандартное состояние растворенного вещества – это его состояние
врастворе, с концентрацией , равной 1, но сохраняющей свойства разбавленного раствора. Т.е. это не вполне реальное состояние, а полученное искусственно экстраполяцией свойств разбавленного раствора на концентрированный. Идеология такая же как
вописании реальных газов.
µ2
Химический потенциал растворенного вещества по закону Генри
µ2о |
Химический потенциал реального растворенного |
|
вещества |
0 |
ln m2 |
3. Р еальные растворы во всей области концентраций
http://www.mitht.org/forum/

53
Если мы хотим сохранить форму выражения химического потенциала для всей области
существования реалрастворов, то нужно ввести поправки на взаимо-действие компонентов реальных растворов и исправить концентрацию на более надежную величину, которая бы правильно отражала их свойства – такая величина называется активностью.
µ1= µ1 о + R T ln а 1 |
µ2= µ2о + R T ln а 2 |
а = γ х для компонента идеального по Р аулю, стандартное сосотяние - чистое |
|
вещество : γ → 1, когда х → 1 |
|
а = γ m для компонента идеального по Генри |
|
γ → 1, когда m → 0. |
|
13.4.4.Коллигативные свойства разбавленных растворов нелетучих веществ в жидкости.
13.4.4.1. Определение Э то означает, что в паре нет растворенного вещества, и он состоит из одного
растворителя. Кроме того предположим, что в твердой ф азе не образуется твердых ратсворов. Е сли эти условия соблюдены, то разбавленные растворы проявляют ряд свойств, зависящих только от концентрации рсстворенного вещества, но не зависящих от природы растворенного вещества. Э то связано с тем, что свойства этих растворов обусловлены свойствами растворителя, которые, судя по выражению химического потенциала, зависят от его мольной доли, а следовательно, от концентрации растворенного вещества, но не от его природы. К коллигативным свойствам растворов относятся :
понижение температуры замерзания растворителя, повышение температуры кипения раствора по сравнению с чистым ратсворителем; осмотическое давление.
13.4.4.2. Повышение температуры кипения растворителя. Э булиоскопия.
На рассмотренных диаграммах состояния мы видели, что давление пара растворителя над раствором всегда ниже, чем над чистыми веществами, а следовательно температура кипения раствора нелетучих компонентов всегда выше, чем у чистых растворителей. Попробуем описать это явление количественно, т.е. рассмотреть уравнения линий на диаграммах. Линии описывают состояния ф азового равновесия, поэтому будем исходить из его критерия:С овершенно аналогично можно расмотреть равновесие между раствором нелетучего растворенного вещества и его паром.
р |
|
|
|
|
|
|
|
|
|
|
|
µ1(ж)= µ1 о (ж) + R T ln х1 |
= µ1 (пар) |
||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
рвнешн |
|
|
|
|
dp |
dT |
|
|
|
|
µ1(ж) = f (T,x) |
|
|
µ1(пар)= f(T) при р =const |
|||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∂µ |
(ж) |
|
|
Т |
|
∂µ |
(ж) |
|
∂µ |
пар |
||||||||
|
|
||||||||||||||||||||
|
|
|
|
|
|
||||||||||||||||
dµ1 (ж) = |
|
1 |
|
|
dT + |
|
|
1 |
|
dx |
= |
|
|
1 |
|
dT |
|||||
∂Т |
|
∂х |
|
∂Т |
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
х |
|
|
|
|
|
|
|
|
Т |
|
|
|
|
|
|
-S (ж) dT + R T d ln x1 |
= - S (пар)dT, |
|
|
|
|
|
|
|
|||||||||||||
R T d ln x1 |
=- (S (пар) - S (ж.))dT = -∆Н исп. dT./Т кип. , |
при |
|
х1 → 1 ∆Н исп. = ∆Ноисп. |
|||||||||||||||||
d ln (1 – x2 ) = -dx2 = - ∆Ноисп. dT/R T2
x2 = ∆Ноисп. (T кип. - Токип. )/R Токип2 = ∆Ноисп/R Токип2 *∆Т.
http://www.mitht.org/forum/

54
х2 = n2 /n1 = n2 M1 /g1 = m2 M1 /1000.
*∆Т = (R Токип2/∆Ноисп) . x2 = (R Токип2 1000/∆Ноисп удельная )m2 = Кэб. m2
Измерение повышения температуры кипения растворителя в растворе называется эбулиоскопией. Для нее удобно использовать растворители с высокой температурой кипения и низкой теплотой испарения. Измеряя повышения температуры кипения растворителя при растворении в нем определенной навески растворяемого вещества, можно найти моляльную концентрацию его, т.е. рассчитать молярную массу его. Э тот метод называется эбулиоскопическим
m2 = (g2 /M2 )1000/g1 = ∆Т кип/ K эб..
M2 = (g2 / g1 )1000 K эб /∆Т кип.
Э тим методом можно изучать состояние растворенного вещества в растворе, т.е. процессы ассоциации и диссоциации, так как при протекании их эф ф ективная измеренная молекулярная масса будет определяться относительным количеством ассоциированных или диссоциированных молекул по ф ормуле аддитивности.
Мэксп. = МмоноN моно + Мдимер Nдимер. .
Вслучае отклонения ратсворов от идеального вместо моляльности будет стоять
активность растворенного вещества а = γ m. Таким образом методом эбулиоскопии можно находить коэф ф ициенты активности.
13.4.4.3. Понижение температуры замерзания растворителя. Криоскопия.
С овершенно аналогично можно расмотреть равновесие между раствором нелетучего растворенного вещества и твердой ф азой.
µ1 |
(ж)= µ1 о (ж) + R T ln х1 = µ1о (тв.) |
|
dµ1(ж) = dµ1 о (тв.). |
||||||||||||
µ1 |
(ж) = f(T, x), |
|
µ1 о (тв.) = f(T). |
|
|
|
|
о(тв.) |
|||||||
|
|
|
|
∂µ |
(ж ) |
|
∂µ |
(ж ) |
|
|
∂µ |
||||
dµ1 |
(ж) = |
|
|
1 |
|
dT + |
|
1 |
|
dx = |
|
|
1 |
dT |
|
∂Т |
∂х |
|
|
∂Т |
|||||||||||
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
х |
|
|
|
Т |
|
|
|
|
|
-S (ж) dT + R T d ln x1 = - S o(тв.)dT,
R T d ln x1 |
= (S (ж) - S o(тв.))dT = ∆Н раствdT . /Т пл.. , при х1 → 1 |
∆Н раств = ∆Нопл. |
d ln x1 = ∆Нопл dT/R T2 |
|
|
х |
Т |
|
∫d ln x 1 = ∫∆Нопл.dT / R T 2 ln x1 = ln(1-x2 ) ≈ -x2 |
= ∆Нопл /R (1/Tопл. - 1/Т пл. ). |
|
1 |
То |
|
-x2 = ∆Нопл (T пл. - Топл. )/R Топл 2 = ∆Нопл /R Топл 2 *∆Т
-∆Т замерз.1 = R Топл 2/∆Нопл * х2 = m2 R Топл 2*1000/∆Нопл удельная = K кр. m2
х2 = n2 /n1 = n2 M1 /g1 = m2 M1 /1000.
K кр называется криоскопической константой и зависит только от свойств растворителя.
Э тим методом можно изучать состояние растворенного вещества в растворе, т.е. процессы ассоциации и диссоциации, так как при протекании их эф ф ективная измеренная молекулярная масса будет определяться относительным количеством ассоциированных или диссоциированных молекул по ф ормуле аддитивности.
Мэксп. = МмоноN моно + Мдимер Nдимер. .
Вещество |
tкипо С |
tпл о С |
Кэб. , K . кг/моль |
Ккр. , K . кг/моль |
Уксусная кисл. |
118 |
17,2 |
2,93 |
3,9 |
Бензол |
80 |
5,4 |
2,53 |
5,12 |
Вода |
100 |
0 |
0,51 |
1,86 |
|
|
|
|
|
Камф ора |
|
|
|
40 |
http://www.mitht.org/forum/
55
13.4.4.4. Уравнение Шредера, растворимость твердых веществ в жидкости.
Хотя растворимость не относится к коллигативным свойствам растворов, но общность подхода позволяет рассмотреть этот вопрос в этом разделе.
µ2 |
(ж)= µ2 о (ж) + R T ln х2 = µ2о (тв.) |
|
dµ2(ж) = dµ2 о (тв.). |
||||||||||||
µ1 |
(ж) = f(T, x), |
|
µ1 о (тв.) = f(T). |
|
|
|
|
о(тв.) |
|||||||
|
|
|
|
∂µ |
(ж ) |
|
∂µ |
(ж ) |
|
|
∂µ |
||||
dµ2 |
(ж) = |
|
|
2 |
|
dT + |
|
2 |
|
dx = |
|
|
2 |
dT |
|
∂Т |
∂х |
|
|
∂Т |
|||||||||||
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
х |
|
|
|
Т |
|
|
|
|
|
-S (ж) dT + R T d ln x2 = - S o(тв.)dT,
R T d ln x2 = (S (ж) - S o(тв.))dT = ∆Н раствdT . /Т пл.. , при х2 → 1 ∆Н раств = ∆Нопл.
d ln x2 = ∆Нопл dT/R T2
х |
Т |
опл.dT / R T 2 |
∫d ln x 2 = ∫∆Н 2 |
||
1 |
То |
|
ln x2 = |
∆Нопл /R (1/T опл. - 1/Т пл. )= (∆Нопл /R Топл )(T пл. - Топл ) |
|
T пл. = Топл + (R Топл 2 /∆Нопл )ln x2
Для разбавленных растворов замена ∆Н раств = ∆Нопл. оправдана, однако при увеличении концентрации 2-го компонента нужно использовать в уравнении Шредера истинные диф ф еренциальные теплоты растворения.
13.4.4.5. Я вление осмоса. Осмотическое давление
раствор
∆h
растворитель
http://www.mitht.org/forum/

53
Лекция № 17
12.6.4. Уравнение Шредера, растворимость твердых веществ в жидкости.
Хотя растворимость не относится к коллигативным свойствам растворов, но общность подхода позволяет рассмотреть этот вопрос в этом разделе.
µ2(ж)= µ2о (ж) + RT ln х2 = µ2о (тв.) |
|
|
|
dµ2(ж) = dµ2о (тв.). |
|||||||
|
µ1(ж) = f(T, x), |
|
µ1о (тв.) = f(T). |
|
|||||||
|
|
∂µ (ж) |
|
|
∂µ (ж) |
|
∂µ о(тв.) |
|
|||
dµ2 |
(ж) = |
2 |
|
dT + |
2 |
|
dx = |
2 |
dT |
||
|
|
|
|
||||||||
|
|
∂Т |
|
|
∂х |
|
|
|
∂Т |
|
|
|
|
х |
|
|
Т |
|
|
||||
|
-S(ж) dT + RT d ln x2 = - So(тв.)dT, |
|
|||||||||
RT d ln x2 = (S(ж) - So(тв.))dT = ∆НраствdT./Тпл.., |
при х2 → 1 ∆Нраств = ∆Нопл. |
||||||||||
|
|
d ln x2 = ∆НоплdT/RT2 |
|
|
|||||||
|
|
х |
|
Т |
|
|
|
|
|
|
|
|
|
∫d ln x2 |
= ∫∆Н2опл.dT / RT2 |
|
|||||||
|
|
1 |
|
То |
|
|
|
|
|
|
|
ln x2 = |
∆Нопл/R(1/T опл.- 1/Тпл.)= |
(∆Н |
о |
пл/R Топл)(Tпл.- Топл) |
|||||||
|
Tпл.= Топл + (R Топл2 |
/∆Н |
о |
пл )ln x2 |
|
||||||
Для разбавленных растворов замена ∆Нраств |
= ∆Нопл. оправдана, однако при увеличении |
||||||||||
концентрации 2-го компонента нужно использовать в уравнении Шредера истинные дифференциальные теплоты растворения.
12.7. Методы определения коэффициентов активности компонентов раствора
12.7.1. Прямые методы связаны с определением характеристик того вещества, для которого определяется активность.
12.7.1.1. По парциальному давлению пара компонента Коэффициент активности, характеризующий отклонение от идеальности, равен отноше-
нию парциального давления компонента над реальным раствором к давлению над идеальным раствором (по закону Рауля или закону Генри)
Р |
Т = const |
fk |
pk |
|
роВ |
аk = fko = pko |
|
Рреал
При низких давлениях летучесть можно заменить на давле-
|
Рид.. |
ние. |
|
аk = γk xk; γk = ak /xk = pk/ pko xk |
|
|
|
|
АВ
12.7.1.2.По распределению вещества между двумя жидкими фазами.
12.7.1.3.По распределению вещества между жидкой и поверхностной фазой (адсорбция)
13.7.2.Косвенные методы. В этих методах измеряют характеристики 2-го ком-
понента раствора. В основе этих методов лежит уравнение Гиббса-Дюгема:
|
|
SdT – V dp + Σnkdµk = 0/ |
|
При Т,р =const это уравнение принимает вид |
|
|
|
Σ nkdµk = 0 Σ Nkdµk = 0; Σ Nk RT d lnNk = 0; Σ xk d lnxk = 0; |
-x2/ x1 |
= |
d lnx1/ d lnx2; уравнение Гиббса-Дюгема-Маргулеса для идеальных растворов; |
-x2/ x1 |
= |
d lna1/ d lna2; уравнение Гиббса-Дюгема-Маргулеса-Льюиса для реальных растворов. |
Активность растворителя можно определять по активности растворенного вещества, используя коллигативные свойства растворов, т.е. вместо моляльности в формулах нужно использовать активность.
13. Несмешивающиеся и частично смешивающиеся жидкости.
13.1. Термодинамика смешения.
Чтобы жидкости смешивались, необходимо, чтобы ∆Gсмеш. < 0; т.е. ∆Н - Т∆S < 0. Для идеальных растворов это условие выполняется всегда, но в случае отклонений от закона Рауля возможно обратное соотношение.
http://www.mitht.org/forum/

54
13.1.1. Положительное отклонение от закона Рауля.
∆Нсмеш. > 0; ∆Sсмеш. > 0; ∆Gсмеш. > 0, если ∆Hсмеш. > T ∆Sсмеш.
∆Gсмеш. < 0, если ∆Hсмеш. < T ∆Sсмеш
С повышением температуры возрастает роль энтропийного фактора, т.е. уменьшается область расслоения и увеличивается область смешиваемости.
13.1.2. Отрицательные отклонения от закона Рауля
∆Нсмеш. < 0; ∆Sсмеш. < 0; ∆Gсмеш. < 0, если ∆Hсмеш. > T ∆Sсмеш. ∆Gсмеш. > 0, если ∆Hсмеш. < T ∆Sсмеш
С повышением температуры область смешения сокращается.
13.2.Диаграммы состояния частично смешивающихся жидкостей ниже температуры кипения.
Положительное отклонение |
Отрицательное отклонение |
||||||||||||||
Т |
|
|
р=const |
|
Т |
|
|
р=const |
|
|
|||||
|
|
|
|
||||||||||||
|
|
Раствор |
|
|
|
|
|
|
|
υ=1 |
|
|
|||
|
|
υ =2 |
Ткрит. |
|
|
α |
|
β |
|
|
|||||
|
|
υ=1 |
|
|
|
|
|
α + β |
|
Ткрит |
|
|
|||
|
|
|
|
α |
|
|
β |
|
|
|
|
|
|
|
|
|
α+β |
|
|
|
|
|
|
Раствор |
υ =2 |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
А |
|
|
|
NB |
B |
|
|
|
|
NB |
|
B |
|
|
|
|
|
|
|
A |
|
|
|||||||
|
|
Гексан |
|
|
|
Анилин |
|
Вода |
|
|
Анилин |
||||
13.3.Равновесие жидкость - пар
13.3.1.Диаграммы кипения несмешивающихся жидкостей. Перегонка с водяным па-
ром.
Диаграммы имеют такой же вид, как диаграммы плавкости с простой эвтектикой, так как в конденсированной фазе находятся чистые компоненты.
Т,оС |
|
|
р= const |
|
|
|
100 |
робщ. = робенз. + ровода |
|
|
|
|
|
||||||
|
|
|
|
|
|||||
|
|
|
Пар |
|
|
|
|
робенз.< робщ. > ровода |
|
|
|
|
υ = 2 |
υ = 1 |
|
||||
|
|
|
пар + |
|
|
80 |
Токип. бенз.> Ткип. смеси < Токип вода |
||
|
|
|
|
|
|||||
пар + бензол |
|
ж |
|
водаж |
|
|
|
||
|
|
|
А |
|
|
|
68 |
убенз./увода = ро |
бенз./ровода = n бенз./nвода |
|
|
бензолж + водаж υ = |
1 |
n = g/M; g1 |
/g2 = po1M1/po2 M2 |
||||
Бензол |
|
|
Вода |
|
|
||||
Такая диаграмма служит основой для очистки высококипящих веществ методом перегонки с водяным паром. Для этого в перегонный куб пропускают водяной пар, который увлекает в пар чистый компонент, а затем конденсат расслаивается. Это позволяет понизить температуру кипения, что важно, если вещество кипит при высокой температуре и может разлагаться при этом. Точка А на диаграмме называется гетероазеотропной точкой.
13.3.2. Частично смешивающиеся жидкости
Диаграммы кипения частично смешивающихся жидкостей получаются комбинацией диаграмм кипения растворов и диаграмм расслоения двух растворов. При этом диаграм-
мы кипения могут быть без азеотропных точек и с ними. |
|
|
|
|
Токип. В |
|||||||||||
Т |
|
р = const |
|
|
|
Т |
|
р = const |
|
|
||||||
|
Пар + |
Пар |
|
Токип. A |
|
Пар |
υ= 2 |
υ= 1 |
|
|||||||
|
Раствор α |
|
|
|
|
|
Пар+ |
|
|
Пар+ |
|
|||||
|
|
|
υ = 1 |
|
|
|
|
|
α |
|
|
β |
|
|
||
|
α |
С |
|
Dпар + |
F |
Токип. В |
|
|
α C |
Е |
D |
β |
|
|||
|
υ =2 α +β |
β β |
|
|
|
|
α + β |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A |
|
|
|
NB |
B |
|
A |
|
|
|
NB |
|
B |
|||
|
|
|
|
|
|
|
|
|||||||||
http://www.mitht.org/forum/
55
Вода – фенол, метанол – тетраэтилсилан, |
Вода – диэтилкетон, вода – анилин. |
гексан – анилин, бензол – ацетамид |
|
po1 > pCD > po2; T окип 1 < ТСD < Tокип. 2 |
po1 < pCD > po2; T окип 1 > ТСD < Tокип. 2 |
Гетероазеотропная смесь кипит при постоянной температуре и имеет одинаковый состав пара и жидкости.
13.4. Распределение вещества между двумя несмешивающимися жидкостями. Экстракция
|
|
T = const |
|
µ J2 бензол =µ J2вода |
|
|
|||
|
|
J2 |
|
µо бензол + R T ln aбензол = µо вода + R T ln aвода |
|
|
|
µо бензол - µо вода = f(T) = RT ln aвода/абензол = -RT ln K |
|
|
Бензол |
|
||
|
|
|
|
K – константа распределения |
|
|
|
|
|
|
Вода |
|
К = exp(-∆µ°/RT) |
|
В разбавленных растворах константа распределения может быть выражена через отно-
шение концентраций. Е сли в одном растворе концентрация мала, а в другом вели-
ка, то появляется возможность определять коэф ф ициенты активности.
К = mб/mв при m → 0; K = γ б mб / mв γ в = 1.
Распределение веществ между двумя несмешивающимися жидкостями лежит в основе метода разделения смесей на компоненты, называемого экстракцией, который все шире применяется и в органической и неорганической химии. В настоящее время гидрометаллургия вытесняет пирометаллургические процессы.
14. Растворы электролитов
14.1.Основные понятия
В80-х годах 19 века появилась теория электролитической диссоциации Аррениуса, которая объясняла свойства некоторых растворов проводить электрический ток диссоциацией растворенного вещества на ионы при взаимодействии его с растворителем.
Вещества, способные образовывать ионные растворы или расплавы, называются электролитами. Электролиты делятся на сильные и слабые. Четкой границы между ними нет. В разных средах вещество может вести себя и как сильный, и как слабый электролит.
Слабые электролиты имеют низкую степень диссоциации
http://www.mitht.org/forum/