

Rogers Computational Chemistry Using the PC
.pdf214 |
COMPUTATIONAL CHEMISTRY USING THE PC |
calculated and the direction of the dipole is correct. We shall treat more refined dipole moment calculations in later chapters.
Chemical Application: Bond Orders
Just as it is possible to calculate the electron probability densities at carbon atoms in a p system, so it is possible to calculate the probability densities between atoms. These calculations bear a rough quantitative relationship to the chemical bonds connecting atoms. Bond orders have been correlated with bond lengths and vibrational force constants. Because we are calculating only p electron densities, the results relate only to p bonds. As the reader may anticipate from the discussion to this point, bonds are not localized between atom pairs in MO theory but are delocalized over the entire p system. One can use Mathcad, either of the MOBAS programs, or the simple Huckel molecular orbital program SHMO (see section on programs below) to obtain the eigenfunctions and eigenvalues for butadiene shown in Fig. 7-2.
The definition of bond order is
X
Pjk ¼ Naijaik ð7-33Þ
where the summation is over all occupied orbitals connecting atoms Cj and Ck, N is the number of electrons in a single orbital (1 or 2), and aij and aik are the coefficients of atoms Cj and Ck . The symbol Pjk is given the name bond order; it is a measure of the probability of finding a p electron between atoms Cj and Ck. If Pjk is large relative to the other bond orders, we anticipate a strong bond. In later
|
π |
α−1.62β |
|
|
|
||
α |
π |
α−0.62β |
|
π |
|||
|
α+0.62β |
||
|
|
π |
α+1.62β |
|
01
|
|
0 |
1 |
0 |
0 |
|
H : |
¼ B |
1 |
0 |
1 |
0 |
|
0 |
1 |
0 |
1 |
|
||
|
|
|||||
|
0 |
0 |
1 |
0 C |
||
|
B |
|
|
|
|
C |
|
@ |
|
|
|
|
A |
|
|
|
|
0 |
0:618 |
1 |
|
|
|
|
0 |
0:602 |
0:372 |
0:602 |
0:372 |
1 |
eigenvals |
ð |
H |
Þ ¼ |
1:618 |
eigenveces |
ð |
H |
Þ ¼ |
0:372 |
0:602 |
0:372 |
0:602 |
||||
|
|
B |
0:618 |
|
|
|
B |
0:372 |
0:602 |
0:372 |
0:602 |
C |
||||
|
|
|
|
1:618 C |
|
|
|
|
0:602 |
0:372 |
0:602 |
0:372 |
||||
|
|
|
|
B |
|
C |
|
|
|
|
B |
|
|
|
|
C |
|
|
|
|
@ |
|
A |
|
|
|
|
@ |
|
|
|
A |
|
Energy-Level Spectrum for 1,3-Butadiene.

HUCKEL MOLECULAR ORBITAL THEORY II: EIGENVECTORS |
215 |
discussions, the term population will also be used for this symbol to denote the relative expected electron density.
For buta-1,3-diene, the order of the 1,2 bond is
Pjk ¼ P12 ¼ 2ð0:372Þð0:602Þ þ 2ð0:602Þð0:372Þ ¼ 0:896
(Note that the order of internal nodes in the eigenvectors in Fig. 7-2 is 1, 0, 2, and 3 from left to right.)
In the case of the p bond of ethylene, or any isolated p bond, the bond order is 1.0. We may take the value 0.896 for the bond order at the 1,2 position as an indication that the 1,2 p bond in buta-1,3-diene is not exactly the same as the p bond in ethylene ðP12 ¼ 1:00Þ but is somewhat diminished by delocalization of electrons over the molecular orbital system. Adding the single s bond to this result, the ‘‘double bond’’ in 1,3-butadiene is really a ‘‘1.896 bond.’’ This delocalization of electrons away from the isolated double bond in the 1,2 position implies an augmentation of the bond order in the 2,3 position; it ought to be more than a single bond by the electron probability density gained from the terminal bonds. The summation of bond orders is not necessarily the same as the number of bonds. For example, in Huckel theory, buta-1,3-diene with 3 s and 2 p bonds has a total bond order that is 5.23.
Exercise 7-3
Calculate the bond orders for the 2,3 and 3,4 bonds in butadiene. Is the 2,3 bond augmented at the expense of the terminal bonds?
Solution 7-3
C C C C
1.891.45 1.89 Yes
Chemical Application: Delocalization Energy
We have already obtained solutions for localized ground-state ethylene leading to the energy E ¼ 2a þ 2b. In looking at allyl, the next more complicated case, we can regard it as an isolated double bond between two sp2 carbons to which an sp3 carbon is attached,
C C C
or, in valence bond terminology, a resonance hybrid
C C C $ C C C

216 |
COMPUTATIONAL CHEMISTRY USING THE PC |
In molecular orbital terminology, the hybrid might be represented by one structure with delocalized p electrons spread over the sigma-bonded framework
C C C
These latter two descriptions are equivalent. Chemical evidence leads us to accept either the valence bond or the molecular orbital representation as preferable to the localized representation. One can calculate the eigenvalue (energy) of the delocalized system and the localized system. Taking a ¼ 0 as a reference point, the localized double bond has an energy of 2b but the delocalized model has E ¼ 2:828b because it has 2 electrons in the bonding orbital at 1.414b (Exercise 7-1). The difference between the two, 0.828b, is the delocalization energy of allyl. This is the Huckel molecular orbital equivalent of the experimentally observed stabilization energy of a double bond by a methyl group.
Exercise 7- 4
Write the secular matrix for localized buta-1,3-diene
CH2 CHCH CH2
Solution 7-4
01
|
x |
1 |
0 |
0 |
|
|
|
|
B |
1 |
x |
0 |
0 |
|
ð |
|
Þ |
0 |
0 |
1 |
x C |
7-34 |
||||
B |
0 |
0 |
x |
1 |
C |
|
|
|
@ |
|
|
|
|
A |
|
|
|
The matrix is the 1,3-butadiene matrix with the ones representing the atoms C2 and C3 omitted to reflect the localized nature of the two terminal p bonds. Another way of looking at this matrix is to regard it as representing two ethylene (localized) p bonds in the same linear molecule.
Exercise 7-5
Calculate the delocalization energies of the positive ion, free radical, and negative ion of the allyl model.
Solution 7-5
The energy of the isolated double bond is 2a þ 2b. Both ions and the free radical of the allyl system have eigenvalue energies of 2a þ 2:828b. The difference is 0.828b in all three cases; hence, the delocalization energies are all 0.828b. The reason the energy is not changed by adding an electron to the allyl positive ion to obtain the free radical is that the electron goes into a nonbonding orbital, which neither augments nor diminishes the


HUCKEL MOLECULAR ORBITAL THEORY II: EIGENVECTORS |
219 |
||
LIBRARY PROJECT 7-1 |
j |
The History of |
|
|
|
Resonance and Aromaticity |
|
Write an essay of approximately 2000 words on the history and the various definitions of the concept of resonance, resonance energy, and aromaticity.
Valence-bond theory explains the stabilization of systems of conjugated double bonds (like those in buta-1,3-diene) with resonance structures that are analogous to the resonating oscillators in the section on normal modes of motion in Chapter 5 (see also Wheland, 1955). This is the origin of the term resonance stabilization. The analogy must not be carried too far, however, because energy does not pass from one resonance structure to another as it does from one harmonic oscillator to another. Resonance extremes are hypothetical structures used to describe the unique real molecule. The difference between the enthalpy of hydrogenation anticipated on the basis of a single hypothetical reference structure and the experimental enthalpy of hydrogenation of the real molecule is one definition of the theoretical resonance energy.
We have used the term resonance energy in the heading of this section largely to connect with the older literature. Because of the different ways resonance energy can be defined and the somewhat contentious literature surrounding the term, the more general term stabilization energy has come into the molecular orbital literature and is preferable. In this somewhat less restrictive terminology, buta- 1,3-diene is said to be stabilized relative to some reference standard. Just as molecules can be stabilized by electronic interactions, they can also be destabilized and a destabilization energy can be measured relative, once again, to an arbitrary standard state.
Exercise 7-6
Turner (1957) measured hydH298 of cyclohexene and found it to be 113 kJ mol 1. Using cyclohexene as the reference standard, calculate the resonance energies of
cyclohexa-1,3-diene ( hydH298 ¼ 224 kJ mol 1), cyclohexa-1,4-diene ( hydH298 ¼225 kJ mol 1) and benzene ( hydH298 ¼ 216 kJ mol 1). Comment on these results.
The value for benzene (Kistiakowsky, 1938) has been corrected to conform with the experimental conditions of Turner’s results.
Extended Huckel Theory---Wheland’s Method
One restriction imposed by Huckel theory that is rather easy to release is that of zero overlap for nearest-neighbor interactions. One can retain a E as the diagonal elements in the secular matrix and replace b by b EjS as nearest-neighbor elements where S is the overlap integral. Now,
0 b EjS a Ej |
b EjS |
|
1 |
7-36 |
||
B |
a Ej |
b EjS |
|
0 |
C |
ð Þ |
0 |
|
b EjS a |
Ej |
|||
B |
|
|
|
|
C |
|
@ |
|
|
|
|
A |
|
is the secular matrix.

220 |
COMPUTATIONAL CHEMISTRY USING THE PC |
By analogy to the substitution x ¼ a E=b in the section on the secular matrix in Chapter 6, we can make the substitution
x |
a Ej |
|
7-37 |
|
|
¼ b EjS |
ð |
Þ |
|||
|
|
which causes the secular matrix to take the same form that it did in the simple Huckel theory, for example,
|
1 |
x |
ð6-44Þ |
||||
|
|
x |
1 |
|
|
|
|
for ethylene. From the definition of x, |
|
|
|
||||
E |
j ¼ |
a xb |
ð |
7-38 |
Þ |
||
1 xS |
|||||||
|
|
||||||
Millikan has shown that the overlap integral for hydrogen-like p orbitals in linear hydrocarbons is about 0.27 (Millikan, 1949).
Exercise 7-7
Prove Eq. (7-38) from the definition of x.
Exercise 7-8
What is the energy separation E2 E1 of the bonding and antibonding orbitals in ethylene, assuming that the overlap integral S is 0.27?
Solution 7-8
The eigenfunctions are 1 as in the simple Huckel calculation for ethylene
a þ b
E1 ¼ 1:27
a b
E2 ¼ 0:73
where, by convention, we have chosen E1 < E2 . If we take
b ¼ 1; and a ¼ 0
this leads to
Ej ¼ 0:79; 1:37 ð j ¼ 1; 2Þ
The separation is 2.16b, which, considering all the approximations already made, is not greatly different from the separation we found ignoring overlap.
HUCKEL MOLECULAR ORBITAL THEORY II: EIGENVECTORS |
221 |
Extended Huckel Theory---Hoffman’s EHT Method
Hoffman’s extended Huckel theory, EHT (Hoffman, 1963), includes all bonding orbitals in the secular matrix rather than just all p bonding orbitals. This inclusion increases the complexity of the calculations so that they are not practical without a computer. The basis set is a linear combination that includes only valence orbitals
X
cj ¼ aijfi ð7-39Þ
but even for ethylene, this leads to a 12 12 secular matrix because there are 4 valence electrons on each of 2 carbon atoms and 1 on each of the 4 hydrogens.
The orbitals used for methane, for example, are four 1s Slater orbitals of hydrogen and one 2s and three 2p Slater orbitals of carbon, leading to an 8 8 secular matrix. Slater orbitals are systematic approximations to atomic orbitals that are widely used in computer applications. We will investigate Slater orbitals in more detail in later chapters.
We fill the secular matrix H with elements Hij over the entire set of valence orbitals. The diagonal elements are
13.6 |
1s |
Hydrogen |
Hii ¼ 21:4 |
2s |
Carbon |
11.4 |
2p |
Carbon |
which are the atomic ionization energies in electron volts (eV). The EHT energies are negative as always for bound states relative to an arbitrary zero of energy defined as the energy of the unbound state. (See Computer Project 3-3 for determination of the ionization energy of hydrogen.)
Off-diagonals are given by
Hij ¼ 0:88ðHii þ HjjÞSij
where Sij is, once again, the overlap integral. The off-diagonal Hij is the arithmetic mean of Hii and Hjj modified by the overlap integral and multiplied by another empirical factor, 1:75=2 ¼ 0:88. The Sij are obtained from the Slater orbitals, which closely resemble atomic orbitals in shape. The sum of the occupied EHT orbital energies times 2 (for 2 electrons per orbital in the nondegenerate ground state) is the total energy of the valence electrons in the molecule relative to a reference state of the completely ionized core atoms. The core of C is its nucleus plus its 1s electrons.
In the process of diagonalization, the trace of the EHT matrix is invariant but the spacing between energy levels changes. Changes in spacing are brought about by the presence of nonzero off-diagonal matrix elements. In methane, for example, the energy of the lowest 4 EHT orbitals gets lower and the energy of the highest 4 orbitals goes up by an equal amount.
222 |
COMPUTATIONAL CHEMISTRY USING THE PC |
Exercise 7-9
Sum Hii for the EHT matrix of methane and so obtain the trace of the EHT matrix.
Solution 7-9
X
HiiðC; 4HÞ ¼ 110 eV
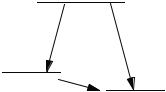
After diagonalization of the EHT matrix, the lowest 4 orbitals have an energy sum of about 70 eV. The electronic energy for these doubly occupied orbitals is 2ð 70Þ ¼140 eV. The energy gain of the molecule relative to its atoms is 140 ð 110Þ ¼30 eV ¼ 690 kcal mol 1 (1 eV 23 kcal mol 1); therefore, the molecule is stable relative to its atoms. We can envision an energy cycle with three steps (Fig. 7-5):
A. Completely independent core positive ions and electrons come together from infinite separation to form 1 C atom and 4 H atoms. One electron per atomic orbital brings about a total energy change of 110 eV.
B. Completely independent core positive ions and electrons come together to form 1 CH4 molecule. Two electrons per occupied bonding orbital bring about an energy change of about 140 eV.
C. The energy of formation of CH4 in the gaseous state from the gaseous atoms is found from the difference
CðgÞ þ 4 HðgÞ ! CH4ðgÞ
By this scheme, the energy of formation of methane from its gaseous independent atoms is about 30 eV ¼ 690 kcal mol 1. This is not the thermodynamic energy of formation in the standard state as it is usually defined but is closely related to it. The energy of formation defined in this way is the reverse of the energy of atomization of methane. Results of molecular orbital calculations are often reported as atomization energies, particularly in the literature before 1990. The EHT value is quantitatively wrong (the experimental value is 390 kcal mol 1), but the method can be indefinitely improved by self-consistent field iteration (Chapter 8) and reparameterization.
The Sij are geometry dependent; hence, one can try various molecular geometries and select the structure that gives the lowest energy, thereby obtaining the best geometry from among the alternatives tried. We have already seen this procedure used in molecular mechanics. It is a concept that will be used at a sophisticated level in the remaining chapters of this book. Hoffman makes the point, richly confirmed by subsequent studies,
|
|
Core ions |
|
–110 eV |
–140 eV |
|
|
|
|
Core atoms |
|
|
–30 eV |
Methane |
Figure 7-5 EHT Energy Diagram for Methane. |
|
|
|
|