

Глава 2
КАК УСТАНАВЛИВАЮТ СТРУКТУРЫ
ОБЩИЙ ВЗГЛЯД
В руках у исследователя неизвестный полисахарид (не будем говорить о том, как он был выделен и очищен – это само по себе большая и сложная тема). Белый порошок, растворим в воде, нерастворим в обычных органических растворителях. Вот, собственно, и все, что о нем пока известно. А что нужно узнать?Структуру. Иными словами, «расставить по местам» те десятки тысяч атомов, из которых состоят молекулы;связать их одним единственным способом ковалентными связями. В последней фразе задача сформулирована вполне точно, однако решить такую задачу «в лоб» современной науке не под силу. Нельзя последовательно установить положение одного атома за другим, если общее их число измеряется тысячами или десятками тысяч – это потребовало бы невообразимых затрат труда и времени*. Поэтому общая стратугия структурного анализа таких сложных объектов состоит в «разборке» молекулы на более мелкие блоки, установлении структуры этих блоков (если и они сложны, то также путем предварительного расщепления на еще более мелкие фрагменты) и затем в реконструкции (мысленной) исходной системы. К счастью (и это далеко не случайное везение, а глубоко обоснованный биологический принцип), все биополимеры построены именно по блочному типу и по самой своей природе сравнительно легко допускают такую разборку. Это значит, что в их молекулах чередуются сравнительно легко расщепляемые связи и участки из значительно более прочных связей. Такие участки и есть те самые блоки,
на которые естественно расщепляются макромолекулы биополимеров.
В полисахаридах легко расщепляемые связи – гликозидные. Разрыв всех гликозидных связей в полисахариде приводит к образованию моносахаридов, из остатков которых был построен полисахарид. Разрыв части гликозидных связей ведет к получению более крупных фрагментов, например олигосахаридов. После того, как установлена структура единичных блоков – моносахаридов (что является относительно простой задачей), структурный анализ исходной системы состоит уже в расстановке не десятков тысяч или тысяч атомов, а немногих тысяч или немногих сотен моносахаридных остатков по определенным местам – задача, все еще весьма сложная, но уже разрешимая. Для ее решения надо узнать, каким путем (из множества возможных) эти мономеры были соединены в полисахаридной молекулы, выяснить размеры циклов моносахаридных остатков (пиранозные или фуранозные) в исходной цепи и установить конфигурации их гликозидных связей.
Задача установления строения смешанных биополимеров гораздо сложнее. Она включает установление строения полисахаридных цепей как одну из подчиненных задач. А в целом надо еще узнать природу и структуру неуглеводной части молекулы, способ присоединения одной части к другой и места присоединения. Так, для установления полной структуры рассмотренных выше группоспецифичных гликопротеинов необходимо узнать структуру полисахаридных цепей, способ, с помощью которого они связаны с полипептидной цепью, структуру узлов связи, структуру полипептидной цепи и, наконец, места присоединения в этой цепи. Это весьма значительная по объему работа. Не случайно после двух десятилетий интенсивных усилий нескольких крупных лабораторий мира полная структура этих биополимеров все еще не установлена (хотя ее основные черты и многие детали уже известны).
Структурные методы в химии углеводов развивались и совершенствовались на протяжении многих десятилетий и сейчас составляют богатый арсенал. В этой части книги мы остановимся на наиболее важных, принципиальных методах, которые реально применяются в самых передовых современных исследованиях и не будем останавливаться на исторических аспектах и на методах и принципах,
уходящих в прошлое, хотя все еще и находящих применение. Мы сделаем лишь одно отступление от этого принципа изложения, но предмет, о котором при этом расскажем, того заслуживает.
Ограничимся разбором путей установления структуры полисахаридов, хотя они далеко не исчерпывают структурные задачи, возникающие в химии углеводов. Для этого есть две причины. Во-первых, полисахариды (включая сюда смешанные биополимеры) представляют собой наиболее важный объект углеводной химии. Во-вторых, установление строения полисахаридов включает основные типы структурных задач, в том числе установление строения моно- и олигосахаридов, а применяемые для этой цели методы являются наиболее общими и употребительными инструментами химии сахаров в целом.
Мономерный анализ
В химии полимеров мономерным анализом называют выяснение вопроса о том, из каких мономерных остатков построен изучаемый полимер. В химии полисахаридов мономерный анализ должен прежде всего установить, из каких моносахаридов построен полисахарид. Для этого нужно расщепить его до моносахаридов, т.е. разорвать все гликозидные связи. Важнейшая реакция, с помощью которой такой результат может быть достигнут,- это кислотный гидролиз гликозидных связей, представленный на примере гидролиза фрагмента -13-связанногоD-глюкана.

Скорости гидролиза гликозидных связей варьируют достаточно широко в зависимости от природы гликозидного остатка, конфигурации гликозидной связи и особенно сильно – от размера цикла. Для пиранозных звеньев обычных альдоз примерные условия полного гидролиза:1н. Минеральная кислота,100С, несколько часов. Для фуранозных звеньев: 0,01н. минеральная кислота, 100С, 1-2 часа.
После гидролиза можно выделить образовавшиеся моносахариды, установить их строение и таким образом узнать, каков моносахаридный состав полисахарида. Конечно, знание моносахаридного состава не позволяет сделать никаких заключений о последовательности моносахаридных остатков в цепи, о регулярности или нерегулярности ее структуры*, о наличии или отсутствии разветвлений – словом, ни об одной характеристике макромолекулы как целого. В этом смысле его можно уподобить данным элементного анализа низкомолекулярного вещества. Более того, моносахаридный состав полисахарида умалчивает даже о многих особенностях строения самых моносахаридных остатков в полисахаридной цепи.
В самом деле, гидролиз осуществляют в сильнокислых средах. Поэтому образовавшиеся моносахариды мгновенно (или во всяком случае неизмеримо быстрее, чем идет гидролиз) достигают мутаротационного равновесия. Таким образом, каков бы ни был размер цикла моносахаридного остатка в полисахаридной цепи и какова бы ни была конфигурация его гликозидной связи, образующийся моносахарид будет получен в одной и той же форме – равновесной смеси, состав которой определяется только условиями среды, а отнюдь не структурой соответствующего звена в полисахаридной цепи. Иными словами, вся информация о размере цикла и о конфигурации гликозидной связи будет необратимо потеряна в результате гидролиза.
Далее. Гидролиз глюкана, который мы привели выше, дает D-глюкозу. Тот же результат получился бы и при гидролизе целлюлозы, и при гидролизе амилозы, глюкана овса или вообще любого другогоD-глюкана. Между тем именно мономерные звенья в этих полисахаридах различаются весьма сильно – не только конфигурацией гликозидных связей, но и местами присоединения остатков друг к другу. В этом смысле истинные мономеры названных полисахаридов – неD-глюкоза, а- или-D-глюкопираноза со связями13или14. Но простой кислотный гидролиз не позволяет их различить. Чтобы после гидролиза не потерять информацию о положении гликозидных связей в исходном полисахариде, надо предварительно
как-то «пометить» атомы кислорода, использованные для образования гликозидной связи и для образования циклов. Непосредственно сделать это нельзя (по крайней мере современными средствами). Но можно решить почти эквивалентную обратную задачу:«пометить» все те кислородные атомы моносахаридного остатка, которые не использованы для образования гликозидных связей и циклов. Это достигается методом метилирования.
Спиртовые гидроксилы можно превратить в простые эфиры, как и всякие спирты. Простейшая возможность – метиловые эфиры. Для этого полисахарид надо обработать теми или иными метилирующими агентами (например иодистым метилом) – прометилировать. Идея метода заключается в том, что метиловые эфиры сахаров устойчивы в условиях кислотного гидролиза гликозидных связей. Поэтому после гидролиза метилового эфира полисахарида можно получить метиловые эфиры входящих в его состав моносахаридов, причем метильные группы в них окажутся в тех же самых положениях, в которых они были в соответствующих моносахаридных остатках полисахаридной цепи. Напротив, неметилированными в них будут те гидроксилы, которые были использованы для образования гликозидных связей и освободились при гидролизе. Таким образом, установив строение метилированных моносахаридов и, следовательно, положение в них метильных групп, можно выяснить, какими своими положениями эти моносахариды были связаны в исходной полисахаридной цепи. Все это можно проследить на примере метилирования растворимого ламинарина, фрагмент которого представлен на схеме (с. 53).
Метилирование полисахарида приводит к образованию его полного метилового эфира, не содержащего свободных гидроксильных групп. Последующий гидролиз дает смесь частично метилированных глюкоз (1-3).
Тетраметиловый эфир 1 не содержит свободных спиртовых гидроксилов и, следовательно, происходит из концевых остатков – тех, которые были в полисахариде только гликозильными. Поскольку в этом соединении в положении 4 имеется метильная группа, этот гидроксил не мог быть включен в цикл в исходном остатке.
Следовательно, этот остаток был не фуранозным, а пиранозным.
Триметиловый эфир 2 содержит один свободный спиртовый гидроксил в положении 3. Следовательно, эти
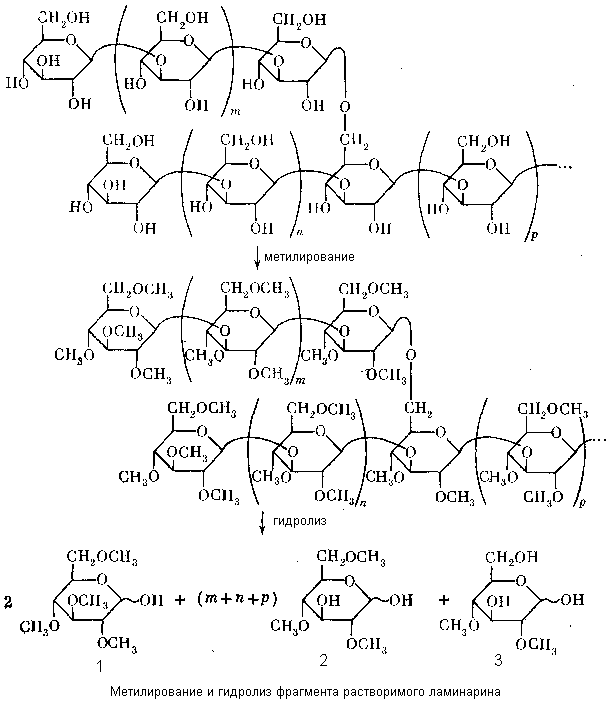
остатки находились внутри цепи и были привязаны к соседнему с ними гликозильному остатку (слева, при традиционной форме записи полисахаридных структур) связями 13. Наличие метильной группы при О-4 указывает на то, что эти остатки находились в полисахариде в пиранозной форме.
Диметиловый эфир 3 содержит два свободных спиртовых гидроксила. Из этого прямо следует, что к этому остатку в полисахариде было присоединено два гликозильных, т.е. что он служил местом разветвления (а значит, исходный полисахарид был разветвленный). А положение свободных гидроксилов указывает на положение их связей 13 и16. Иными словами, становится ясно, что полисахаридная цепь построенная из13-связанных остатков, разветвлена по положениям 6.
Таким образом, видно, что метод метилирования позволяет выполнить гораздо более детальный мономерный анализ полисахарида, установить не только природу моносахаридных остатков, из которых он построен, но и положения межмономерных связей в каждом остатке и даже тип структуры (разветвленный-неразветвленный). Следует, однако, помнить, что при всех своих достоинствах метод метилирования не есть прямой способ установить, какие атомы кислорода вовлечены в межмономерные связи и циклы. Это лишь метод, основанный на рассуждении от противного («поскольку этот гидроксил метилирован, он не был использован для образования гликозидных связей или циклов. Следовательно …»). А при таком способе могут возникать неопределенности. Мы не будем их здесь разбирать, а хотим только предупредить читателя об абсолютизации этого метода (рассуждение:«делали метилирование, значит есть структура», довольно распространено) и отослать его за подробностями к более специальной литературе.
Количественный анализ состава смеси метилированных моносахаридов, полученных из полисахарида, позволяет установить также среднуюю длину линейных участков цепи, или среднюю частоту разветвлений (по соотношению продуктов типа 2 и 3), а также оценить среднюю молекулярную массу полисахарида по соотношению продуктов типа 2 и 1 (для неразветвленных полисахаридов)*.
Мы видим, что метилирование – высоко информативный метод структурного анализа полисахаридов. Но тем не менее это всего лишь метод мономерного анализа, который в принципе, по самой сути, не может дать представления
о полной структуре полисахарида – последовательности звеньев, распределении остатков между различными участками цепей (например, в рассмотренном примере с ламинарином о распределении остатков между главной и боковой цепями), о конфигурации гликозидных связей. Здесь буквально реализуется та самая потеря исходной части связей, по поводу которой так зло иронизировал гётевский Мефистофель:
«…живой предмет желая изучить
Чтоб ясное о нем познанье получить
Ученый прежде душу изгоняет
Затем предмет на части расчленяет
И видит их, да жаль духовная их связь
Тем временем исчезла, унеслась!»*
Для восстановления утраченных характеристик структуры полисахаридов нужны принципиально другие методы, которые мы разберем в последующих главах. Но сначала надо рассказать о том, как устанавливают строение моносахаридов и их метиловых эфиров, т.е. о структурной концовке мономерного анализа.
УСТАНОВЛЕНИЕ СТРОЕНИЯ МОНОСАХАРИДОВ
Идентификация
Итак, осуществлен гидролиз полисахарида и получены составляющие его моносахариды или метилированные сахара. Теперь надо установить их строение. Задача эта все еще достаточно сложна и трудоемка (хотя и проще, чем установление строения самого полисахарида). Поэтому, прежде чем непосредственно браться за ее решение, следует подумать, нельзя ли установить строение …, не занимаясь установлением строения?Часто оказывается, что можно. В арсенале органической химии есть такой прием, который позволяет прийти к определенным выводам о структуре молекулы без ее последовательной экспериментальной расшифровки. Этот прием называется идентификацией вещества.
Научный потенциал, накопленный человечеством к сегодняшнему дню, состоит не только в огромных общих знаниях, могущественных методах исследования и совершенных приборах. В него входят также сведения о точных, хорошо воспроизводимых физических характеристиках гигантского числа органических соединений, в том числе моносахаридов и их производных, строение которых уже было установлено определенно и надежно. И если нам удастся доказать, что полученный из неизвестного полисахарида моносахарид тождествен или, как чаще говорят, идентичен известному моносахариду, мы тем самым установим строение этого моносахарида. Доказательство идентичности двух веществ – идентификация – есть один из важнейших во всей органической химии принципов исследования, а применяемые для этой цели методы и приемы постоянно совершенствуются и развиваются. Как же практически идентифицировать органическое соединение, в частности моносахарид?
Прежде всего можно определить его физические константы. Самые обычные и легко измеряемые из них – температура плавления и удельное вращение. После этого пора обратиться к литературе – не был ли описан ранее моносахарид с такими константами?И если окажется, что был описан, у исследователя появляется, нет,не уверенность, но только основание для предположения о том, что его моносахарид идентичен известному, и, следовательно, право предположительно приписать ему определенную структуру. Почему же только предположительно?А вот почему.
Прежде всего, точность определения этих констант и их воспроизводимость сравнительно невелики. Реально обе эти величины могут быть измерены с помощью обычных приборов с ошибкой, достигающей 1-2. Сами же величины зависят от чистоты образца, а она может быть различной у нашего исследователя и у того, кто впервые описал это вещество в литературе. Так что даже для безусловно идентичных веществ расхождение в температурах плавления и величинах удельного вращения вполне может достигать 2-3. Температуры плавления моносахаридов, например, лежат обычно в интервале примерно 50-200*. При допустимой ошибке в 3это означает всего
около 50 различных температур плавления. Иными словами, вероятность случайного совпадения этой константы – порядка 2%. Это, конечно, недопустимо много. Не намного лучше обстоит дело с удельным вращением. Для большинства моносахаридов удельные вращенияумещаются в интервале от -100до +100, т.е. вероятность случайного совпадения удельного вращения разных соединений опыть около 1-2%. Таким образом, даже совпадение обеих этих констант для двух моносахаридов может оказаться случайным с вероятностью в несколько сотых процента. Такая вероятность ошибочного вывода об идентичности все еще велика, для того чтобы серьезный ученый мог ею удовлетвориться*. Ко всему прочему нужно добавить, что очень многие моносахариды, а особенно метилированные сахара весьма трудно получить в кристаллическом состоянии, причем сделать это тем труднее, чем хуже очищено вещество и чем меньшим его количеством располагает исследователь. А если нет кристаллов, то нельзя и определить температуру плавления.
Остаются еще, конечно, и другие характеристики вещества:его спектры, цветные реакции, некоторые особенности химического поведения. Однако, во-первых, они обычно менее индивидуальны и характерны для данного соединения, во-вторых, далеко не для всех соединений с известной структурой описаны в литературе (в отличие от двух самых распространенных:температуры плавления и удельного вращения). Таким образом, «заочная» идентификация соединения по литературным данным – вещь мало надежная. Совсем другое дело – держатьв руках два образца:неизвестного вещества и известного, устроить им, так сказать, очную ставку. В научной литературе это называется «идентифицировать вещество путем прямого сравнения с заведомым образцом». Здесь возможности для надежной идентификации резко расширяются.
Прежде всего, существует общая закономерность:два разных вещества могут случайно иметь одинаковые
температуры плавления, но их смесь обязательно будет плавиться при другой температуре. Только смесь двух идентичных веществ (которую в сущности вообще нельзя назвать смесью, разве что смесью двух образцов) имеет точно такую же температуру плавления, что и исходные компоненты. Так что отсутствие депрессии температуры плавления – чрезвычайно простой и очень надежный метод идентификации двух веществ при прямом сравнении. Для этого, однако, нужно иметь чистое кристаллическое вещество, а это не всегда удается.
В таком случае вступает в свои права комплекс мощных аналитических методов – хроматография. Это способ анализа веществ, основанных на их физическом разделения. Например, при хроматографии на бумаге вещества двигаются по хроматограмме с током растворителя с различными скоростями, индивидуальными и характерными для данного вещества в данных условиях. Последняя оговорка весьма существенна:в разных лабораториях и в разных руках точно воспроизвести абсолютные скорости – их называют хроматографическими подвижностями – весьма и весьма трудно. Поэтому здесь не обойтись литературными данными – нужно прямое сравнение двух образцов.
Огромное достоинство хроматографических методов в том, что они позволяют работать с очень малыми количествами вещества (например, порядка микрограмма) и, что еще важнее, позволяют идентифицировать не только мало очищенные вещества, но даже вещества, присутствующие в качестве компонентов сложных смесей. Последнее особенно существенно для разбираемой нами задачи, так как, например, гидролизат полисахарида может содержать несколько разных моносахаридов. И хроматография позволяет идентифицировать их без предварительного разделения.
Конечно, и хроматографические методы могут дать осечку:подвижности разных веществ могут и случайно совпасть. Однако хроматография – это очень гибкий метод. Можно использовать набор разных условий для анализа одной и той же пары веществ,а совпадение подвижностей в нескольких различных условиях – это уже событие, вероятность которого ничтожно мала*.
Кажется, все?Проблема идентификации моносахаридов решена? Не тут-то было! В одном и очень важном для химии углеводов пункте хроматографические методы бессильны:они не позволяют отличать правое от левого. Мы можем, например, со всей надежностью идентифицировать хроматографически галактозу, но останемся в полном неведении относительно того,D- или L-галактозу мы имеем.
Значит, нужно сделать еще что-то. Тут два пути. Можно выделить моносахарид (или его производное) в индивидуальном состоянии и определить его удельное вращение. А можно воспользоваться ферментом, катализирующим ту или иную реакцию этого моносахарида – уж ферменты-то отличают правое от левого! Но ферменты – реагенты тонкие и капризные. Надежный анализ с помощью ферментативной реакции требует проверки с применением образцов заведомых моносахаридов, вводимых в ту же реакцию с темже самым препаратом фермента. И вот тогда только у исследователя появляется действительная уверенность в том, что вещество – моносахарид – идентифицировано.
Мы видели, какую большую роль при идентификации играет прямое сравнение с заведомым образцом. Спрашивается, а где его взять? Промышленность реактивов выпускает специальные наборы образцов многих природных веществ, и в частности моносахаридов (разумеется, самых обычных и распространенных). Но это минимум, который гораздо ниже «прожиточного» при развитой исследовательской работе. Поэтому каждый исследователь, а также исследовательский коллектив, стремятся создать свою собственную коллекцию образцов веществ, пополняют ее при любой возможности и берегут как зеницу ока. Поэтому, в частности, столь значительно повышается эффективность исследовательской работы в больших коллективах:многие проблемы легко решаются путем обмена образцами известных веществ. Наконец, образцы известных соединений являются предметом международного обмена и сотрудничества. Неудивительно,
что ученые часто получают письма примерно такого содержания:«Глубокоуважаемый докторN! Не могли бы вы прислать нам несколько миллиграммов такого-то вещества для идентификации?». И в ответ летят в авиаконвертах через океаны и континенты считанное число драгоценных кристалликов. Поэтому научная статья нередко кончается словами: «Авторы глубоко признательны докторуN, любезно предоставившему заведомый образец такого-то вещества». И тут есть за что благодарить: иной раз один такой кристаллик позволяет исследователю сэкономить многие месяцы, а то и годы труда.
Историческое отступление
Мы обещали касаться только вполне современных методов исследования, и не без оснований:классику легко найти в любом учебнике*. И все-таки хочется отсупить от этого принципа и описать методы, с помощью которых были впервые выяснены конфигурации ассиметрических центров важнейших моносахаридов. Это – классическая работа Эмиля Фишера**. Изложим ее несколько упрощенно, стремясь сохранить главное – логику исследования.
Прежде всего о том, что уже было известно Фишеру и что предстояло узнать. Была известна бутлеровская структура (см. с. 8) нескольких моносахаридов, было также известно, что для некоторых из них возможны взаимные превращения путем определенных реакций и что изомерные альдозы отличаются конфигурацией ассиметричемких
центров. Установить же нужно было относительные конфигурации этих центров. Экспериментально Фишер использовал главным образом две реакции.
Циангидриновый синтез, т.е. присоединение к альдозам синильной кислоты с последующим гидролизом нитрильной группы и восстановлением карбоксильной до альдегидной. Результатом этой реакции является удлинение углеродной цепи альдозы на одно звено и возникновение нового ассиметрического центра, вследствие чего из каждого моносахарида получается не один, а два новых моносахарида – стереоизомеров поC-2. В общем виде эта последовательность показана на схеме:

Окисление азотной кислотой. При этом и альдегидная группа, и концевоеCH2OH-звено окисляются до карбонильных групп. В результате молекула приобретает повышенную симметрию – происходит уравнивание концов, причем в зависимости от относительной конфигурации ассиметрических центров эта симметрия может оказаться полной или неполной. Ниже эта реакция показана на примереD-галактозы:

Фишер располагал тремя альдозами:глюкозой, маннозой и арабинозой. Для последней, если оперировать с сахарамиD-ряда, принципиально возможны четыре конфигурации(4-7):

После окисления арабинозы азотной кислотой Фишер получил оптически деятельную, следовательно ассиметричную дикарбоновую кислоту. Это сразу исключало две структуры из четырех возможных. В самом деле, ожидаемые продукты из четырех возможных пентоз следующие (8-11):

Первые две из этих кислот обладают плоскостью симметрии (указана на схеме пунктиром) и, значит, должны быть оптически неактивны. Таким образом, этот эксперимент оставлял для кислоты только две возможные структуры: 10 и 11, а для арабинозы – только 6 и 7.
Путем циангидринного синтеза из арабинозы были получены две гексозы: глюкоза и манноза. Из этого следовало, что у последних конфигурация центров приC-3, C-4иC-5такая же, как в арабинозе (у ееC-2, C-3иC-4соответственно), и что различия между глюкозой и маннозой сводятся только к различию конфигурацииC-2. Таким образом, глюкозе и маннозе могут соответствовать либо пара конфигураций 12 и 13 (если арабиноза- это 6), либо пара 14 и 15 (если арабиноза – это 7).

Для выбора между этими парами Фишер опять применил принцип уравнивания концов. Четырем гексозам 12-15 соответствуют четыре дикарбоновые кислоты (16-19):

Из этих четырех кислот только одна – 18 – обладает плоскостью симметрии и, следовательно, оптически недеятельна. Окислив глюкозу и маннозу, Фишер получил две оптически активные кислоты. Таким образом, глюкозе и маннозе не может соответствовать пара конфигураций 14 и 15, а соответствует только пара 12 и 13. Следовательно, для арабинозы (из которой, как мы помним, могут быть получены эти две гексозы) остается только одна возможная структура – 6.
Теперь Фишер уже знал, что глюкоза и манноза имеют конфигурацию 12 и 13, но еще не знал, какая из этих конфигураций отвечает глюкозе, а какая маннозе. Вопрос был решен аналогичным образом:последовательным применением циангидринного синтеза и окисления, которые приводили к образованиюC7-дикарбоновых кислот. Из маннозы при этом были получены две оптически активные кислоты, что возможно только при исходной конфигурации 13 (читатель может сам вывести их структуры и доказать асимметрию), а из глюкозы – две кислоты
одна из которых была оптически недеятельна. Такая симметричная структура – кислота 20 –могла возникнуть только из гексозы с конфигурацией 12. Таким образом, и последний вопрос этой серии – конфигурация глюкозы и маннозы – был блестяще разрешен Фишером.

Позднее аналогичным образом он установил относительные конфигурации остальных основных пентоз и гексоз и тем самым впервые создал научную основу всей химии углеводов.
Работа Фишера, опубликованная в 1891 году, даже по сегодняшним меркам должна быть оценена как первоклассное исследование – по безукоризненной логике, тщательности экспериментального выполнения и полной строгости и надежности заключений. В настоящее время асимметрия насыщенного углеродного атома есть не вызывающая сомнений школьная истина. Однако во времена Фишера это была лишь сравнительно недавно сформулированная (Вант-Гоффом и Ле Белем в 1874 г.) стереохимическая гипотеза, имевшая очень немного экспериментальных подтверждений даже для очень простых систем, содержащих один-два ассиметрических атома. Нужна была глубокая убежденность в ее справедливости, глубокая уверенность в применимости строгой логики к сложным органическим соединениям и в надежности и однозначности превращений, чтобы задумать, предпринять и блестяще довести до конца такое (кстати сказать, экспериментально весьма сложное) исследование. Поэтому работу Эмиля Фишера по установлению конфигурации моносахаридов смело можно отнести к истинно гениальным творениям, которые не только приводят к блистательным конкретным результатам, но и освещают путь своим глубоким идейным содержанием новые пути в целой области науки. Начиная с этой работы
стереохимическая гипотеза превратилась в стереохимическую теорию – одно из наиболее фундаментальных обобщений органической химии.
Логический приемы, введенные Фишером, широко используются в структурных исследованиях до сих пор (правда, на иной химической основе). Приведем здесь только два примера, в основу которых положено уравнивание концов:сведениеD-арабинозы (21) иD-ликсозы(7) к одному и тому же полиолу (22), и идентификацию 3,4-ди-О-метил-D-ксилозы (23), для которой заведомый образец труднодоступен, в виде полиола 24 с его оптическим антиподом 25, который образуется из более доступной 2,3-ди-О-метил-D-ксилозы (26):

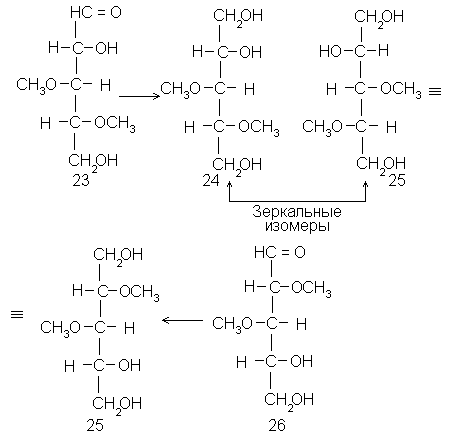
В обоих примерах для перехода от альдоз к полиолам использовано восстановление боргидридом натрия. Видно, что в обоих случаях, при всем внешнем несходстве, применена стереохимическая логика, заимствованная из работы Фишера.
Теперь поясним, как устанавливают структуру и конфигурации моносахаридов и их метилированных производных в современных работах. Здесь решающую роль играют два метода – осколочная масс-спектрометрия для установления структур (без стереохимии) и спектроскопия ядерного магнитного резонанса (ЯМР) для выяснения конфигураций ассиметрических центров.
Масс-спектрометрия
Итак, имеется моносахарид или его метилированное производное. Установить строение – значит решить две группы задач. Прежде всего надо выяснить длину углеродной цепи, природу, число и расположение функциональных групп;для метилированных сахаров, в частности,- число и положение метильных групп. Все это в совокупности иногда называют бутлеровской структурой. Затем нужно установить конфигурацию ассиметрических центров, т.е. решить задачу того же типа, которую решал Эмиль Фишер для глюкозы, маннозы и арабинозы. В этой главе мы рассмотрим пути решения задач первой группы одним наиболее общим и употребительным в современной науке методом – с помощью осколочной масс-спектрометрии.
Принципиально масс-спектрометр состоит из четырех блоков:системы напуска, ионного источника, системы магнитной фокусировки и детектора (рис. 1). В системе напуска образец анализируемого вещества испаряют в вакууме. Образовавшиеся пары поступают в ионный источник, где подвергаются бомбардировке пучком ускоренных электронов (энергия обычно порядка десятков электронвольт). Энергия облучения расходуется на выбивание электронов из молекул анализируемого вещества – последние превращаются в положительно заряженные ион-радикалы. Такие частицы высоко реакционноспособны и нестойки. Тут же в ионизационной камеры они претерпевают распад на заряженные и незаряженные осколки (отсюда название метода «осколочная масс-спектрометрия»). Вся ионизационная камера находится под высоким положительным

потенциалом по отношению к остальным частям прибора. Поэтому электростатическое поле выталкивает из камеры положительные ионы. Перед выходом из камеры пучок ионов проходит через систему электростатических линз и диафрагм, так что в результате из камеры выходит узкий сфокусированный ионный луч, в котором скорости ионов зависят от их масс и зарядов.
Ионные пучок далее попадает в зону магнитной фокусировки. Здесь в магнитном поле прямолинейные траектории ионов искривляются, причем геометрия магнитного поля рассчитана так, чтобы сфокусировать ионы на детекторе. В конечном итоге ионы подходят к детектору по индивидуальным траекториям, которые целиком определяются величиной отношения массы иона к его заряду (m/e). Варьируя электростатическое или магнитное поле, можно сфокусировать на детекторе ионные потоки для каждого значенияm/eи измерить количественно соответствующий таким частицам ионный ток, т.е. величину, пропорциональную числу частиц с даннымm/eв анализируемой плазме. Развертка поm/eдает масс-спектр, в котором по оси абсцисс отложены величиныm/e, а по оси ординат – интенсивности ионного тока, или, что то же самое, доля частиц с даннымm/e в плазме (рис. 2). Поскольку в подавляющем большинстве случаев образующиеся осколки однозарядны, шкалаm/eпрактически совпадает со шкалой ионных масс.
В описанных условиях масс-спектрометрия (а они самые обычные, но далеко не единственные) органические

вещества дают сложные масс-спектры. В них, однако, удается выделить наиболее характерные и наиболее интенсивные пики, отвечающие главным путям распада изучаемого соединения. Поскольку типичные пути распада многих классов органических соединений, в частности моносахаридов, сейчас подробно изучены, по картине масс-спектра можно составить достаточно полное представление о структуре изучаемого соединения, затратив на это минимум вещества (меньше миллиграмма, нередко лишь микрограммы) и минимум времени (на съемку спектра на хороших приборах требуются считанные минуты;иное дело, что расшифровка спектра может занять несравненно больше времени).
Как же расшифровывают масс-спектры?«Читают» спектр обычно справа налево – от больших масс к малым. И это не прихоть:крупные осколки обычно наиболее информативны. Для них возможно лишь весьма ограниченное число путей образования, тогда как «мелочь» может возникать самыми разными путями и из нее извлечь аналитически полезную информацию гораздо труднее.
Первый пик в спектре – пик молекулярного иона, т.е. ионизированной, но не распавшейся исходной молекулы*. При описании спектра его обозначают буквой М. Уже из этого пика можно извлечь много полезных сведений. В самом деле, молекулярные массы – это не температуры плавления или удельные вращения. Они могут иметь только дискретные значения, подчиняющиеся
вполне определенным закономерностям. Ну, например, таким простейшим, как то, что любое соединение состава CnHmOp может иметь только четную молекулярную массу. Значение молекулярной массы сразу резко ограничивает число возможных структур, а более подробный анализ спектра в области пика молекулярного иона позволяет получить еще целый ряд дополнительных данных. Мы здесь не будем разбирать этот аспект, а укажем лишь на один характериный пример. Природный бром состоит из двух изотопов79Brи81Brв соотношении 1:1. Поэтому молекулярный ион любого соединения, содержащего один атом брома, дает в масс спектре два пика равной интенсивности, различающиеся на две единицы массы. Такой дуплет в спектре весьма характерен и сразу указывает на наличие в анализируемом соединении одного атома брома. А если бы в нем было два атома брома, то соответствующие ионы дали бы пик в виде триплета с расстоянием между компонентами в две единицы массы и соотношением интенсивностей1:2:1.
Дальше по спектру идут пики осколков. Молекулярный ион распадается на две частицы: заряженную и нейтральную. Последняя часто оказывается высокоустойчивой малой молекулой типаH2O, CO и т.п. Эти фрагменты нейтральны, однако их можно идентифицировать косвенно – по разности масс молекулярного иона и заряженного осколка. Поэтому последние часто описывают в разностном выражении, например:M-H2O, или M-18; M-CO, или M-28; M-CH3, илиM-15; M-H2C=C=O, или М-42 и т.д. Состав таких больших осколков обычно легко идентифицировать, так как число вариантов состава малых осколков весьма невелико. Так, например, для обычных органических соединений М-18 – это всегдаM-H2O*. Таких первичных
осколков, т.е. тех, которые возникают непосредственно за распадом молекулярного иона, может быть несколько, так как распад может протекать по нескольким направлениям. Первичные осколки, в свою очередь, подвергаются распаду, а продукты распада тоже распадаются. Так возникают серии ионов, отвечающих определенным путям распада, или, как чаще говорят, фрагментации молекулярного иона. Искусство расшифровки спектра в значительной мере состоит в умении из большого числа пиков выделить такие, которые увязываются в определенные серии – последовательности фрагментации исходного иона. Когда такие серии выявлены, восстановить картину распада и, следовательно, структуру анализируемого вещества уже значительно проще, особенно если исследователь опирается на общие данные о характерных путях фрагментации соединений данного класса.
Теперь, наконец, можно уже конкретно перейти к масс-спектроскопии моносахаридов. Непосредственно исследовать их этим методов затруднительно. Дело в том, что молекулы моносахаридов содержат много полярных групп, а это самым неблагоприятным образом сказывается на их летучести. Выход из положения состоит в получении подходящих более летучих производных. На их выбор накладывается целый ряд ограничений, но к настоящему времени эта трудность уже преодолена:найдено несколько классов производных, отвечающих всем требованиям, и подробно изучены закономерности их фрагментации. Чаще всего для этой цели сейчас используются ацетаты полиолов. Их получают с помощью двух весьма общих и чрезвычайно простых в экспериментальном оформлении реакций: восстановление моносахарида боргидридом натрия и последуюшего ацетилирования. Ниже эти реакции показаны на примереD-галактозы (с. 71).
Фрагментация ацетатов полиолов такого типа относительно проста. Она включает первичные ионы, образующиеся путем разрыва C-Cсвязей углеродного скелета, а также отщепленияCH3COO. Зная массу таких осколков и массу молекулярного иона, легко составить представление

о структуре исходного полиола и, следовательно, моносахарида. Если в молекуле имеется дезоксизвено, как например в ацетате 28, образующемся из 2-дезокси-D-рибозы (27), то характерным оказывается разрыв цепи вβ-положении к дезоксизвену. Зная эту закономерность, по величинеm/eсоответствующих фрагментов (в данном случаеm/e 159) можно установить положение дезоксизвена (см. с. 72).
Тут, однако, возникает одна неопределенность. Действительно, ацетат 29, образующийся из изомерной 4-дезоксирибозы 30, будет давать точно такой же фрагмент с m/e159, только из нижней части молекулы. Поэтому мы еще не вправе приписать структуру исходному сахару – это может быть либо 2-дезоксипентоза, либо 4-дезоксипентоза.
Выход, однако, есть. Надо искусственно внести асимметрию в молекулу. Для этого вместо боргидрида натрия на стадии восстановления применяют его изотопный аналог – тетрадейтерийборгидрид. Тогда у углеродного атома бывшей карбонильной группы, т.е. при С-1, появляется один атом дейтерия вместо одного атома протия. В результате ацетат полиола 31 из 2-дезокси-D-рибозы дает в масс спектре тот же характеристический пик, но сдвинутый на единицу массы (разница масс дейтерия и протия), т.е. пик иона сm/e160 вместо 159. А ион, возникающий

путем аналогичной фрагментации из изомерного соединения 32, дейтерия не содержит и, следовательно, будет по прежнему иметь m/e159.
Для метилированных моносахаридов аналогичное превращение дает частично метилированное и частично ацетилированные полиолы. Характерное направление фрагментации таких производных – это разрыв C-C-связи около метоксильной группы. Особенно характерен такой разрыв между двумя соседними метоксильными группами (если такая пара в молекуле имеется):

По таким фрагментам можно установить положение метоксильных групп, т.е. решить главную задачу, возникающую при структурном исследовании метилированных моносахаридов. Неопределенность, возникающая здесь из-за относительно высокой их симметрии (т.е. либо 2,3-ди-О-метил-, либо 4,5-ди-О-метил-) может быть легко устранена аналогично тому, как описано выше, т.е. с применением дейтероборгидрида на стадии восстановления. Вообще надо сказать, что введение изотопной, особенно дейтериевой, метки – весьма распространенный прием в масс-спектрометрии. Вот, например, как была доказана бутлеровская структура 4-О-метил-D-глюкуроновой кислоты, полученной синтетически в виде метилового эфира 33:

Здесь из масс-спектра, точнее, всего из двух пиков ионов с m/e262 и 191, следовала сразу вся структура – число ацетильных и метильных групп, а также тот факт, что в молекулу при восстановлении вошло три атома дейтерия, т.е. что исходное соединение было эфиром гексуроновой кислоты (один дейтерий входит при восстановлении альдегидной группы, а два – при восстановлении этерифицированного карбоксила). Кроме того, пики ионов сm/e191 и 262 однозначно определяют положение метильной группыи и без всяких неопределенностей, так как концы были помечены дейтерием, причем по-разному: у С-1 – один атом, а уC-6 – два.
Итак, масс-спектрометрия – чрезвычайно информативный метод установления строения. Но для нее, конечно, нужно иметь индивидуальное вещество, т.е. произвести предварительное разделение смеси, в которой вещество находится.Такой результат достигается непросто (особенно при работе с метилированными сахарами) требует сложной (и в экспериментальном и в приборном отношении) хроматографической техники. Наивысшее современное достижение в этой области – объединение газо-жидкостного хроматографа и масс-спектрометра в одном приборе, т.е. анализ смеси методом, получившем название хромато-масс-спектрометрии.
В газо-жидкостном хроматографе вещество вносят в колонку – длинную узкую трубку с нелетучей жидкой фазой, нанесенной на пористый инертный твердый материал. Через колонку пропускают струю газа-носителя при определенной регулируемой температуре. Вещество в виде паров движется по колонке с током газа, непрерывно подвергаясь распределению между газовой (подвижной) и жидкой (неподвижной) фазами. Время, в течение которого данное вещество проходит колонку (так называемое время удерживания) зависит от летучести вещества и его способности абсорбироваться данной жидкой фазой. Оба свойства определяются тонкими особенностями структуры конкретного соединения, так что время удерживания весьма характерно и индивидуально для каждого вещества в конкретных условиях разделения. Поэтому, если в колонку внесена смесь веществ, то ее компоненты появляются на выходе из колонки в разное время:достигается их разделения. После выхода их колонки газовый поток попадает в детектор, регистрирующий появление вещества, а сигналы с детектора через усилительную схему поступают
на самописец. В результате самописец выписывает кривые выхода вещества из колонки в зависимости от времени, т.е. рисует газо-жидкостную хроматограмму. Таким образом можно проанализировать состав весьма сложных смесей веществ.
Идея хромато-масс-спектрометрии состоит в том, что газы и пары, прошедшие колонку, вводят в ионизационную камеру масс-спектрометра, объединенного с хроматографом в единый комплекс – хромато-масс-спектрометр. В результате исследователь получает из одного анализа смеси сведения о временах удерживания ее компонентов, об их относительном содержании в смеси и, наконец, тут же получает масс-спектры каждого компонента смеси.
Такой метод анализа идеально подходит для изучения смесей метилированных сахаров, получающихся при мономерном анализе полисахаридов с помощью метода метилирования. В самом деле, хромато-масс-спектрометрия позволяет идентифицировать известные вещества со свидетелями при помощи прямого сравнения и тут же, используя масс-спектрометр, дополнительно подтверждать их структуру, а для неизвестных веществ или для тех, для которых не оказалось нужного заведомого образца,- установить строение (без конфигураций, конечно) по масс-спектру.
В настоящее время хромато-масс-спектрометрия – магистральный путь развития структурного анализа полисахаридов*, позволяющий получить на нескольких миллиграммах изучаемого биополимера за считанные дни информацию, для добывания которой еще совсем недавно требовались десятки, а то и сотни граммов материала и годы труда.
Спектроскопия ЯМР
В отличие от масс-спектрометрии, мы не будем излагать здесь сколько-нибудь подробно физические основы спектроскопии ЯМР. Дело в том, что теория этого метода

и принципы работы приборов значительно сложнее, чем в масс-спектрометрии, а книга наша посвящена прежде всего сахарам. С другой стороны, по спектроскопии ЯМР имеется очень много доступных книг самого различного уровня – от популярных до весьма фундаментальных*. Поэтому мы здесь ограничимся лишь самым поверхностным описанием спектроскопии ядерного магнитного резонанса, причем только резонанса на протонах (ПМР).
В обычном спектре ПМР каждый имеющийся в молекуле протон дает свой сигнал (другое дело, что сигналы двух или более протонов могут накладываться друг на друга). Этот сигнал характеризуется тремя параметрами:интенсивностью, которая прямо пропорциональна содержанию протонов данного типа в образце, величиной химического сдвига, измеряемого от некоторого внутреннего стандарта (чаще всего тетраметилсилана) в единицах м.д. (т.е. в миллионных долях рабочего поля прибора), и величиной константы спин-спинового взаимодействия с другим (или другими) протонами в той же молекуле. Эта константа, проявляющаяся в спектре в виде расщепления сигналов на отдельные компоненты и сокращенно называется обычно КССВ, измеряется в герцах (рис. 3).
Величина химического сдвига определяется многими особенностями структуры органической молекулы, а также рядом внешних факторов (в первую очередь применяемого для съемки спектра растворителя). Наибольший вклад в эту величину вносит плотность электронного облака вокруг данного протона:чем в большей
степени электронная пара, образующая связь данного протона с ближайшим атомом, оттянута к этому последнему, тем большей будет величина химического сдвига δ. В очень грубом приближении можно сказать, что чем более кислым является данный протон, тем больше его химический сдвиг. Поскольку смещение электронов связи определяется в первую очередь природой включенного в эту связь атома, а во вторую – ближайшим окружением этого атома, величина химического сдвига отражает положение данного протона в молекуле и потому находит весьма многообразное применение в установлении структуры органических соединений.
Константа спин-спинового взаимодействия описывает спиновую связь данного протона с другими протонами в той же молекуле. Это взаимодействие осуществляется только по цепи ковалентных связей и поэтому быстро ослабевает с увеличением расстояния между взаимодействующими ядрами. Обычно приходится сталкиваться с КССВ протонов через две связи (в группах H-C-H)и через три связи(в группировкахH-C-C-H). Величины КССВ также определяются многими структурными факторами. Главным из них является геометрия соответствующего фрагмента. Именно на этом обстоятельстве в первую очередь основано применение спектроскопии ПМР для установления конфигурации ассиметрических центров в сахарах. Геометрия фрагментаH-C-H, даже будучи надежно установлена, дает мало интерсных сведений о структуре и конфигурации изучаемого соединения. Поэтому соответствующая ей КССВ играет подчинительную роль в применении ПМР-спектров сахаров (кроме того, таких фрагментов в углеводах всегда мало). Напротив, геометрия фрагментовH-C-C-Hв совокупности определяет всю стереохимию типичной моносахаридной молекулы. Поэтому измерение соответствующих КССВ и их интерпретация играют первостепенную роль в структурном применении спектроскопии ПМР.
Если (в первом приближении) считать длины связей и углы H-C-C иC-C-Hпостоянными величинами, то геометрия фрагментаH-C-C-Hможет быть описана одним параметром:так называемым диэдральным, или двугранным, углом, смысл которого виден из рис. 4.
Величина КССВ двух протонов в фрагменте H-C-C-Hзависит главным образом от диэдрального угла. К сожалению, однако, КССВ зависит не только от этого

угла, но и от ряда других структурных особенностей молекулы. Поэтому все математические описания зависимости КССВ только от угла носят неизбежно приближенный характер (таково, например, широко известное уравнение Карплюса). Тем не менее почти всегда справедливо следующее грубое правило:Если диэдральный угол приближается к 0или 180, то КССВ – «большая», т.е. составляет около 8-10Гц;если этот угол близок к 90, то КССВ близка к 0, если же (и это весьма распространенный случай в реальных структурах) этот угол близок к 60, то КССВ бывает «малая», т.е. порядка 1-3 Гц. Несмотря на явно приближенный характер этого правила, оно находит очень широкое и плодотворное применение в структурных исследованиях. Вот типичный пример из химии углеводов.
Требуется определить конфигурацию гликозидного центра в тетраацетате D-глюкопиранозида, т.е. сделать выбор между двумя структурами:34а и 34б. Анализ основан на КССВ протона при С-1 с протоном при С-2 (этот фрагмент выделен на схеме жирными линиями). В первом случае (α-глюкозид) диэдральный уголH-C-C-Hсоставляет около 60, т.е. для него следует ожидать малой КССВ. Дляβ-аномера (34б) этот угол близок к 180(Это два аксиальных заместителя, их связи параллельны оси цикла). Поэтому для такого соединения КССВ протона при С-1 должна быть большой. Наблюдаемая величина КССВ (8 Гц) позволяет уверенно сделать выбор в пользу структуры 34б.

Внимательный анализ рассмотренного примера показывает, что здесь спектроскопия ПМР позволяет одновременно решить вопрос и о конформации изучаемого соединения, и о конфигурации ассиметрического центра. Точнее говоря, вопрос о конфигурации спектроскопией ПМР в данном случае не решается:она определяется лишь опосредованно, через конформацию. В самом деле, глюкозиду 34б можно, по крайней мере, формально, приписать конформацию 35. В этом случае диэдральный угол фрагментаH-C-C-Hсоставлял бы уже не 180, а около 60, ожидаемая КССВ была бы существенно иной, а выводы из спектра ПМР могли бы оказаться прямо ошибочными. Однако из общих принципов конформационного анализа мы можем заключить, что дляβ-глюкозида конформация 35, если и реализуется, то в достаточно малой пропорции по сравнению с конформацией 34б. Так что сделанное выше заключение остается в силе.
Таким образом, можно видеть, что для эффективного применения спектроскопии ПМР в решении структурных задач в сахарах, т.е. для установления конфигураций, необходимо по возможности уменьшить неопределенность, связанную с возможными конформационными состояниями изучаемых молекул. Прежде всего для этого применяют циклические производные, например гликозиды, и избегают применять ациклические, типа полиолов, в которых конформационные возможности значительно шире. Обычно стремятся применять такие производные и в таких растворителях, для которых конформационные закономерности наиболее просты и однозначны. В этом смысле, например, пиранозные формы сахаров явно предпочтительнее фуранозных, а ацетаты сахаров предпочтительны перед свободными сахарами. К сожалению, дать этому трактовку, не вдаваясь в подробности конформационного анализа, не представляется нам возможным.
В примере с глюкозидами мы показали, что знание
КССВ протона при C-1позволяет сделать надежное заключение о конфигурации ассиметрического центра С-1. И это вполне справедливо при тех условиях задачи, которые мы сформулировали выше:установить аномерную конфигурациюD-глюкопиранозида, т.е. гликозида, для которого известна конфигурация всех остальных центров асимметрии. Однако, если бы мы не знали конфигурации остальных центров, то полученный результат говорил бы лишь об относительной конфигурации двух соседних центров:С-1 и С-2, а не об их абсолютных конфигурациях, что означало бы существование двух вполне равноправных вариантов ответа. Дальнейший анализ спектра показал бы нам, что протон при С-2 имеет, в свою очередь, большую КССВ по сравнению со следующим соседом, последний – протон при С-3 – имеет также большую КССВ с протоном при С-4, а протон при С-4 – большую КССВ с протоном при С-5. Такой набор данных позволил бы заключить, что в изучаемом соединении все протоны пиранозного цикла аксиальны, так как только при этом условии все КССВ этих протонов могли оказаться большими. Это прямо означает, что все заместители в пиранозном цикле экваториальны, т.е. автоматически приводят к глюко-конфигурации (правда, опять относительной:спектроскопия ПМР принципиально не способна отличать оптические антиподы). Таким образом, подробный анализ спектра глюкозида 34б привел бы к четким выводам об относительной конфигурации всех ассиметрических центров, т.е. к решению фишеровской задачи. Именно на таком анализе спектров и основано в первую очередь определение конфигурации моносахаридов с помощью спектроскопии ПМР.
Спектр глюкозида 34б очень прост в том смысле, что все КССВ большие, следовательно все протоны аксиальны. Более того, этот случай уникален. При любой другой конфигурации, какие-то КССВ окажутся малыми, а какие-то большими. Когда нам известно, какая КССВ к какой паре протонов относится, задача решается более или менее просто. Однако на пиках в спектре отнюдь не написано, какому протонув молекуле принадлежит данный сигнал*. Мало того, совсем не исключение, а
скорее правило, что в спектрах производных углеводов сигналы части протонов накладываются друг на друга. Поэтому главной трудностью, с которой сталкивается исследователь при анализе спектра ПМР сахаров, является отнесение сигналов, т.е. установление взаимооднозначного соответствия определенного сигнала в спектре и определенного протона в молекуле. Как же решается такая задача?
Надо сразу сказать, что стандартных путей к решению здесь нет. Точнее говоря, путь есть и совершенно надежный и общий;однако его применение сопряжено с весьма значительными затратами труда и времени. Он состоит в синтезе дейтерированных аналогов изучаемого соединения и сопоставлении спектров аналогов и родоначального вещества. Идея этого подхода основана на том, что дейтерий в спектрах ПМР не виден (его химический сдвиг лежит далеко за пределами шкалы химических сдвигов протонов). В результате в спектре дейтерированного аналога исчезает сигнал одного из протонов, а именно того, который был заменен на дейтерий. И по этому признаку можно с полной надежностью произвести отнесение такого сигнала. Имея серию дейтерированных производных, можно полностью расшифровать весь, сколь угодно сложный, спектр ПМР.
По-видимому, даже непосвященному в таинства органического синтеза ясно, что такой путь очень трудоемок и не может применяться в качестве рутинного метода хотя бы по той простой причине, что для осмысленного планирования синтеза дейтерированных аналогов нужно прежде всего знать структуру соединения, а это лишает смысловой основы расшифровку спектра ПМР как шага на пути установления структуры соединения. Поэтому синтез дейтероаналогов применяется тогда, когда расшифровка сложных спектров имеет самодовлеющее значение, например в исследовании закономерностей спектра ПМР новых классов соединений и т.д. (собственно, закономерности, на которые мы теперь опираемся при структурном примененииПМР, и были в свое время добыты таким трудоемким путем). В рутинном же применении ПМР для структурных исследований отнесение сигналов в значительной мере основывается на изученных ранее особенностях спектров соединений этого класса, на ряде общих закономерностей спектроскопии ПМР, а также на многих частных приемах расшифровки.

Для иллюстрации приведем здесь спектр тетрабензоата α-D-ликсопиранозы (рис. 5) и его трактовку. При этом мы упростим свою задачу: не будем расшифровывать шаг за шагом спектр, полученный с прибора, а попытаемся разобраться, и то лишь в общих чертах, почему сигналы определенных протонов расположились в спектре именно таким образом (будем рассматривать только наиболее информативную часть спектра – протоны пиранозного цикла).
Мы уже упоминали, что главный вклад в величину химического сдвига вносит электронная плотность вокруг данного ядра. В рассматриваемом соединении наименьшая электронная плотность должная окружать протон при С-1. В самом деле, у этого углеродного атома имеется два кислородных, оттягивающих электроны заместителя. Поэтому сигнал протона Н-1 вполне закономерно оказывается в спектре в самом слабом поле, т.е. имеет наибольший химический сдвиг. Наоборот, каждый из протонов при С-5 испытывает влияние другого протона, связанного с тем же углеродным атомом. Связь C-Hполяризована таким образом, что ее электроны несколько смещены к углероду. Поэтому электронная плотность вокруг протонов при С-5 несколько выше, чем вокруг любого другого протона в молекуле. В соответствии с этим оба протона имеют наименьший химический сдвиг (располагаются в наиболее сильном поле). Электронное окружение протонов при С-2, С-3 и С-4 весьма сходно, и неудивительно, что их сигналы располагаются в спектре близко друг к другу. В отнесении этих сигналов могут помочь следующие соображения.
Спин-спиновое взаимодействие двух протонов вполне взаимно:оно приводит к расщеплению сигналов каждого из взаимодействующих протонов в дублет, и, конечно, с одной и той же КССВ. Если с данным протоном связан спиновой связью еще один, то взаимодействие с ним осуществляется независимо от взаимодействия с первым партнером. Таким образом, каждый компонент дублета, возникшего в результате взаимодействия с первым протоном, расщепляется на дублет со свой КССВ. В результате протон, взаимодействующий с двумя другими, дает сигнал в виде квартета (или, точнее, дублета дублетов). Знание этих особенностей (мы снова подчеркиваем, что ни слова не говорим о физике явления, а рассматриваем тот крайний минимум сведений, который можно использовать, так сказать, потребительски) играет большую роль в расшифровке спектров ПМР.Теперь вернемся к нашему примеру.
Протон при С-1 уникален в том отношении, что на расстоянии не более трех ковалентных связей от него находится только один другой протон – при С-2 (у остальных протонов кольца имеется не менее двух таких соседей). Поэтому его сигнал должен быть дублетом, как оно и есть на самом деле. По этому признаку мы могли бы отличить его от всех остальных, даже не прибегая к соображениям химического сдвига. Его взаимодействие с протоном Н-2 описывается КССВ 3,1 Гц. Следовательно, такая же КССВ должна быть у сигнала протона Н-2. Просмотрев остальной спектр, мы обнаруживаем сигнал, у которого одна из КССВ составляет 3,1 Гц и отнесем его к протону при С-2. Измерив его вторую КССВ(3,3 Гц), мы узнем о взаимодействии с протоном при С-3. Аналогично находим и сигнал протона при С-3 (по признаку КССВ 3,3 Гц), и у него обнаруживаем вторую (большую) КССВ (9,0 Гц) с протоном при С-4 и т.д.
Наша задача не в том, чтобы научить читателя методике расшифровке спектров ПМР (в этом смысле изложенное выше весьма схематично), а в том, чтобы по возможности передать логику мышления в этой области. И в связи с этим особенно важно обратить внимание на два обстоятельства. Первое. В наших рассуждениях мы опирались на знаение структуры изучаемого соединения – мы могли не знать его стереохимии, но на бутлеровскую структуру ссылались постоянно. В этом смысле спектроскопия ПМР дает (в некоторых пределах, конечно) тем
больше новых сведений об изучаемом соединении, чем большей информацией о нем исследователь уже располагает. Для изучения структуры «с нуля» метод ПМР часто мало эффективен. Второе. Отнесение сигналов по цепи спиновых связе опирается на отнесение одного (первого) сигнала. Ошибка в первом отнесении неизбежно приведет к полностью ошибочной интерпретации всего спектра. Некоторой гарантией верности отнесений может служить логическая увязка отнесений всех сигналов в спектре как с точки зрения спиновых связей, так и с точки зрения имеющихся сведений о структуре. Поэтому очень рискованно бывает делать серьезные выводы из спектра ПМР, в котором удалось отнести лишь часть сигналов*.
Не вдаваясь в подробности, укажем в заключение, что мощным подспорьем в расшифровке спектров ПМР служит такие инструментальные методы, как двойной резонанс и ИНДОР, позволяющие объективно установить наличие спиновых связей между ядрами;динамический ЯМР (ДЯМР), позмоляющий устранить неопределенности, связанные с конформационными равновесиями, а также метод сдвигающих реагентов (или, как их часто называют на английский манер, шифт-реагентов), с помощью которого можно избирательно и весьма сильно изменить химические сдвиги отдельных протонов (как говорят на лабораторном жаргоне, «вытянуть их из каши», т.е. из группы перекрывающихся и потому почти не поддающихся интерпретации сигналов).
Нам бы не хотелось, чтобы у читателя сложились абсолютистские представления о современных методах установления строения моносахаридов. Ну, например, такие:есть два стандартных метода – масс-спектрометрия и спектроскопия ПМР, которые позволяют автоматически устанавливать строение любых моносахаридов (стоит только им научиться и получить доступ к приборам). Или иначе:без масс-спектров и спектров ПМР структуру не установить. И то и другое неверно.
Конечно, оба эти метода – исключительно мощные инструменты исследования. Однако это отнюдь не «черные ящики», где на входе вещество,а на выходе готовая структура. На выходе – всего лишь спектр, а структура появляется в результате интерпретации спектра. Последняя же отнюдь не трафаретна и требует от исследователя (именно от самого исследователя, а не от того, кто управляет прибором и выдает спектры) больших знаний, опыта, интуиции*. Помимо спектроскопии, современная химия углеводов располагает целым комплексом точных и тонких методов структурного анализа, которые, хотя и не опираются на новейшие приборы, позволяют делать не менее надежные заключения о структуре. Бывает так, что самыми примитивными, известными с прошлого века «пробирочными» пробами можно узнать о структуре моносахарида не меньше, чем используя самую совершенную аппаратуру. Мы, конечно, далеки от того, чтобы пропогандировать идею возврата кэпохе жаровен и реторт, но хотим подчеркнуть широту и многообразие накопленного к настоящему времени арсенала методов структурных исследований. И в оценке той или иной работы самую последнюю роль должны играть соображения новизны примененных методов или, тем более, их модности.
Несравненно важднее, чтобы метод (пришедший ли из прошлого века или из самоновейшей литературы) был применен к месту, чтобы он позволял делать максимально надежные заключения с минимальными затратами труда. И никакой самоновейший метод при бездумном применении не гарантирует от грубых ошибок.
Короче говоря, установление строения моносахаридов как почти любое другое структурное исследование, есть творчество. В нем могут применяться весьма совершенные и разработанные методы исследования, приемы простые и сложные, но их совокупность, последовательность и ход мысли исследователя ни в какие стандартные правила не укладываются
Это и есть творческая деятельность, которая, как известно, отличается от прочих тем, что для нее нельзя составить алгоритма или программы. Так что, в конечном счете, методы суть лишь инструменты, а струтуры устанавливаются не методами, а интеллектуальными усилиями ученых.
УСТАНОВЛЕНИЕ СТРОЕНИЯ ПОЛИСАХАРИДОВ
Что надо узнать?
Возвратимся к вопросу об установлении строения полисахаридов. Мы оставили рассмотрение его на стадии завершения мономерного анализа, включая результаты, полученные методом метилирования. Что же к этому моменту уже известно о структуре, а что еще предстоит узнать?
Известно, из каких моносахаридов построен полисахарид, в какой циклической форме их остатки входят в его состав, каково положение межмономерных связей в остатках каждого типа, каков тип структуры (разветвленный-неразветвленный). Для разветвленных полисахаридов, кроме того, известны степень разветвленности с структура точек ветвления. Это не мало, но это еще не структура. Что же еще не известно?Для всех типов полисахаридов – конфигурация гликозидных связей и последовательность расположения моносахаридных остатков в цепи, а также, за редкими исключениями, молекулярная масса. Для разветвленных полисахаридов к этому еще прибавляется вопрос о распределении остатков между основной и боковыми цепями, о длине боковых цепей и о положении различных точек ветвления (они могут располагаться в главной цепи, в первых от главной боковых цепях, во вторых от главной боковых цепях и т.д.). А для полисахаридов, имеющих неуглеводные заместители, надо еще установить положение этих заместителей. И только для одного – простейшего – типа полисахаридов мономерный анализ дает почти всю структурную информацию – для линейных регулярных полисахаридов, построенных из однотипно связанных остатков одного единственного моносахарида, каковы, например, целлюлоза и амилоза.
Совокупность нерешенных структурных вопросов можно несколько условно разбить на две группы:вопросы
«ближнего» порядка в структуре и вопросы «дальнего порядка». Вот что мы имеем в виду. Выберем в полисахаридной цепи произвольным образом некоторый моносахаридный остаток А. Применяя те или иные методы, о которых речь впереди, можно узнать, с какими другими остатками он связан непосредственно:установить, например, наличие фрагмента …-Б-А-В-… как элемента структуры цепи. Сведения о ближнем порядке моносахаридных остатков в таком отрезке цепи, или, как говорят в полимерной химии, сегменте цепи, сами по себе мало что говорят о структуре всей макромолекулы. Однако знание структуры многих сегментов составляет уже обширную информацию о молекуле в целом. Обширную, но далеко не полную, потому что мы не знаем, как эти сегменты расположить в цепи относительно друг друга, и даже не можем сказать, существуют ли в цепи промежутки неизвестной структуры между известными нам сегментами. Этот вопрос можно отнести к дальнему порядку в структуре цепи, т.е. не о том, что находится на первом, втором и т.д. месте от данного остатка (ближний порядок), а о том, какие остатки располагаются от него в удалении, например, на двадцатом, тридцатом и т.д. месте. Схематически это можно пояснить так:

На схеме окружены примеры элементов ближнего порядка, стрелками указаны примеры элементов дальнего порядка.
Отвлекаясь пока от конкретных методов и приемов исследования, можно сказать, что для исчерпывающей характеристики структуры полисахаридной цепи нужно иметь полную информацию и о ближнем, и о дальнем порядке в макромолекуле. Существующие методы исследования нельзя четко разбить на две группы по характеру получаемого с их помощью ответа о ближнем или дальнем
порядке (так сказать, на методы для ближнего порядка и методы для дальнего порядка). Тем не менее, мы постараемся оценить их информативность именно с этой точки зрения. А рассмотрев важнейшие структурные методы, обсудим сегодняшник возможности установления структуры полисахаридов в целом.
Как узнать?
Общий принцип выяснения последовательности моносахаридных остатков в полисахариде состоит в расщеплении цепи на олигосахаридные фрагменты, установлении строения таких фрагментов и последующей реконструкции структуры макромолекулы по структурам олигосахаридов. Вот два характерных примера применения этого принципа, на которых можно проследить его возможности и ограничения.
В агарозе имеются гликозильные остатки двух видов:β-D-галактопиранозильный и 3,6-ангидро-α-L-галактопиранозильный. Гликозидная связь второго из них гораздо более чувствительна к кислотному гидролизу и некоторым другим аналогичным реакциям расщепления. Поэтому в определенных условиях можно добиться ее избирательного разрыва, не затрагивая при этом гликозидную связь остаткаβ-D-галактопиранозы. В результате полисахаридная цепь распадается на дисахаридные блоки агаробиозы*по схеме, приведенной на с.89.
Образование именно такого дисахарида с полной определенностью указывает на присутствие в исходной цепи отвечающей ему последовательности β-D-галакторираноза-3,6-ангидро-L-галактопираноза, а высокий выход этого дисахарида означает, что такая последовательность доминирует в цепях. Поскольку из мономерного анализа мы знаем, что два вида моносахаридных остатков входят в состав агарозы в соотношении1:1, доминирование такого сегмента возможно только в структуре цепи с чередующимися остатками. Таким образом, мы узнаем о наличии второго типа сегмента:3,6-ангидро-L-галактопираноза-

β-D-галактопираноза*. Схематически это можно представить так:

где Г-остаток β-D-галактопиранозы, А-остаток 3,6-ангидро-α-L-галактопиранозы
В результате анализа полисахарида при помощи такого частичного гидролиза мы получаем, как видно, надежную информацию о ближнем порядке моносахаридных остатков в цепи, т.е. о том, какой остаток с каким связан непосредственно. Мы однако, остаемся в неведении относительно дальнего порядка, что применительно к нашему примеру сводится к вопросу о регулярности строения цепи. Почему же мы не вправе решить его утвердительно на основании приведенных выше данных?Сейчас разберемся.
Если бы избирательность разрыва гликозидных связей при частичном гидролизе была абсолютной, а выход дисахарида – количественным, то из таких результатов можно было бы сделать вполне строгий вывод о регулярности структуры агарозы. Однако даже тогда, когда мы имеем дело с агарозой (случай, совершенно исключительный для всей химии углеводов по степени избирательности химического гидролиза), выход агаробиозы не достигает 100%. Следовательно, мы вправе предполагать наличие в цепи каких-то других последовательностей, помимо тех существование которых мы установили экспериментально. Например, с результатами эксперимена вполне совместимо предположение о наличии некоторого количества сегментов …-А-А-… и …-Г-Г-…, которые по тем или иным причинам не обнаруживают себя при частичном гидролизе:
…-А-Г-А-Г-А-Г-А-А-Г-А-Г-А-Г-А-Г-Г-
-А-Г-А-Г-А-Г-А-Г-…
Правомерность такого предположения не позволяет нам
утверждать, что на основании описанного эксперимента мы установили дальний порядок связей. Мы можем лишь говорить о том, что правильное чередование остатков доминирует в структуре полисахарида*.
Значительное различие в скоростях кислотного гидролиза входящих в полисахарид гликозидных связей – явление сравнительно редкое. Во всяком случае, та степень избирательности, которая достигается для агарозы, уникальна. В типичных же случаях выходы олигосахаридов при частичном гидролизе гораздо ниже и, следовательно, по информативности результатов такие эксперименты значительно уступают рассмотренному примеру.
Другой метод частичного расщепления полисахаридных цепей (деградация по Смиту) основан на более избирательной, но и более сложной последовательности реакций. Ключевой стадией здесь служит окисление полисахарида солями иодной кислоты – периодатами. При этой реакции происходит разрыв С-С-связи в гликолях с образованием диальдегида, последующее восстановление которого приводит к образованию диола, как показано на схеме:

Строго обязательным условием для периодатного окисления является наличие двух гидроксильных групп у соседних углеродных атомов, между которыми и разрывается С-С-связь. Моносахаридное звено в цепи, содержащее систему такого гликоля, расщепляется до ациклического ацеталя, как показано на примереβ-D-глюкопиранозильного звена с заместителем (например, левой частью полисахаридной цепи) в положении 4 (с. 92)
Ациклический ацеталь, образующийся при этих реакциях из окисленного моносахаридного звена, сравнительно легко подвергается кислотному гидролизу, так что последний удается провести высоко избирательно, т.е. не затрагивая

обычные гликозидные связи. В результате происходит разрыв полисахаридной цепи по окисленному звену. При этом левая часть цепи превращается в гликозид многоатомного спирта (в нашем случае эритрита), а в правой появляется концевое моносахаридное звено, которое раньше не было связано с окисленным остатком.
Теперь рассмотрим деградацию по Смиту неразветвленного глюкана из овса, в состав которого входит два типа остатков β-D-глюкопиранозы со связями 13и14. Первое из этих звеньев устойчиво к действию периодата, так как не содержит подходящих гликольных группировок, а окисление второго мы только что обсуждали.
Оценим сначала, каких результатов периодатного окисления для основных типов структуры полисаханида, которые можно умозрительно построить на основании данных мономерного анализа. При правльном чередовании 13- и14-связанных остатков главными продуктами деградации должны быть глюкозил-эритрит и гликолевый альдегид, образующиеся в результате сохранения13-связанного звена и окисления двух примыкающих к нему остатков со связями14 (см. схему на с.93)
При блочном строении цепи, т.е. наличии в ней длинных последовательностей из 13-и14-связанных остатков, первые должны приводить к образованию полимерного неокисленного фрагмента, а вторые – полностью распадаться до свободного эритрита и гликолевого альдегида. Наконец, при хаотическом распределении остатков продуктом деградации должна быть смесь низкомолекулярных гликозил-эритритов, образующихся из коротких последовательностей13-связанных остатков, гликолевого альдегида, а также свободного эритрита, возникающего

при окислении коротких последовательностей остатков со связями 14. Эксперимент показывает что реализуется именно этот третий случай. Действительно, глюкан овса при деградации по Смиту распадается на серию мономерных и олигомерных фрагментов, анализ которых позволяет вывести ряд заключений о его структуре (см. схему)

3Г,4Г – остатки глюкопиранозы, замещенные в положении 3 и 4 соответственно;Г – концевое (незамещенное) глюкопиранозное звено;Э – остаток эритрита или свободный эритрит.
Из того факта, что все содержащие глюкозу фрагменты имеют на восстанавливающем конце остаток эритрита (а не какого-нибудь другого полиола) следует, что в полисахариде существует связь 3Г-4Г. Наличие таких фрагментов,
содержащих два, три и четыре остатка глюкозы, указывает на присутствие в цепи последовательностей из двух, трех и четырех остатков типа 3Г, расположенные подряд. С другой стороны, то, что все продукты деградации низкомолекулярны, свидетельствует об отсутствии в полисахариде длинных последовательностей из таких неокисляемых остатков. Образование свободного эритрита указывает на существование сегментов …-4Г-4Г-…, однако в отличие от последовательностей …-3Г-3Г-… не позволяет сделать никаких заключений об их длине. Определение выходов полученных фрагментов всех типов дает возможность оценить относительное содержание соответствующих сегментов в исходном полисахариде, а их разнообразие указывает на отсутствие простой регулярности в чередовании различных остатков в цепи.
Таким образом, деградация по Смиту позволяет узнать очень многое о ближнем порядке в структуре полисахарида, включая последовательности из нескольких моносахаридных звеньев. Однако о дальнем порядке мы можем сказать еще меньше, чем в случае частичного гидролиза агарозы. Действительно, найденные сегменты цепей могут быть сгруппированы в блоки, могут чередоваться по определенному (достаточно сложному) закону, могут, наконец, распределяться по цепи хаотически. Обо всех этих характеристиках дальнего порядка мы остаемся в неведении.
Два разобранных примера частичного расщепления полисахаридных цепей по информативности характерных для всего цикла методов фрагментации такого типа. С их помощью можно узнать многое или почти все о ближнем порядке в структуре и кое-что или почти ничего о дальнем порядке. Тем не менее, применение к одному полисахариду разных методов фрагментации, в особенности таких, которые обеспечивают расщепление разных связей, весьма существенно обогащает информацию, получаемую каждым из этих методов порознь. Например, если из последовательности …-А-А-Б-В-Г-Д-А-А-Д-Г-Г-Б-… одним методом были получены фрагменты А-А-Б-В-Г, В-Г-Д, Д-Г-Г-Б, а другим – фрагмент Г-Д-А-А-Д-Г, то в совокупности эти данные определяют всю додекасахаридную последовательность, т.е. дают сведения о точной структуре уже довольно значительного сегмента цепи.
Мы еще ничего не сказали о способах определения конфигурации гликозидных связей моносахаридных остатков
в полисахаридных цепях. Между тем этот вопрос нередко оказывается камнем преткновения во всем структурном анализе полисахаридов.
Выяснение конфигурации гликозидных связей – это по существу задача мономерного анализа, так как относится не к характеристике структуры цепей, а к детализации структуры отдельных звеньев. Тем не менее известными сейчас методами мономерного анализа эта задача не решается. Дело в том, что все эти методы по своей сути деструктивны и обязательно включают расщепление гликозидных связей. А при всех известных способах расщепления гликозидных связей, применяемых в мономерном анализе полисахаридов (кроме ферментативного гидролиза, см. ниже), информация о конфигурации этой связи теряется.
Мы уже упоминали, что гликозидный центр вносит наибольший вклад в величину удельного вращения углеводов. Поэтому из величины удельного вращения полисахарида можно сделать некоторые заключения о конфигурации гликозидных связей входящих в него моносахаридных остатков. Однако, поскольку удельное вращение – величина аддитивная*, такие заключения неизбежно носят усредненный характери, т.е. ничего не говорят о том, какие именно остатки имеютα- илиβ-конфигурацию. Лишь в простых и крайних случаях оптическая активность полисахарида позволяет сделать более определенные выводы. Действительно, по величине удельного вращения можно достаточно уверенно сделать заключение о преобладании гликозидных связей с такой-то конфигурацией в полисахаридах, построенных из однотипных моносахаридных остатков с одной конфигурацией гликозидных связей.
Совсем по-другому выглядит задача определения конфигурации гликозидных связей в олигосахаридах, особенно в низших олигосахаридах, получаемых при тех или иных способах фрагментации полисахаридных цепей. Дело в том, что при малом количестве гликозидных связей (в дисахаридах, например, такая связь только одна) удельное вращение уже позволяет с гораздо большей определенностью судить о конфигурации этих связей. Кроме того, в ди-, три-, а при удаче и в тетрасахаридах можно
определить конфигурацию гликозидных центров с помощью спектроскопии ПМР, подобно тому, как это делается в низших гликозидах.
По-видимому, универсальный (гипотетический) метод определения конфигурации гликозидных связей в полисахаридах можно представить себе следующим образом. Это должен быть такой метод расщепления гликозидных связей, который приводил бы количественно к производным моносахаридов, подобно кислотному гидролизу. Но с той, однако, разницей, что структура этих производных должна зависеть от конфигурации расщепляемой гликозидной связи исходного остатка. Тогда мы имели бы метод мономерного анализа, который одновременно давал бы информацию и о природе каждого мономерного звена, и о конфигурации его гликозидной связи. К сожалению, ничем похожим на такое идеальное решение углеводная химия пока не располагает (хотя препятствий принципиального характера к разработке подобного метода не видно). Наилучшее доступное сейчас приближение к идеалу – это окисление ацетатов полисахаридов хромовым ангидридов в уксусной кислоте. Суть этого метода состоит в следующем.
Глюкопиранозильный остаток, гидроксилы которого защищены от окисления ацетилированием, при обработке хромовым ангидридом в уксусной кислоте претерпевает окисление, при котором гликозидная связь превращается в сложноэфирную. Остаток моносахарида превращается при этом в остаток кетоальдоновой кислоты. В эту реакцию вступают только гликозильные остатки, у которых водород при гликозидном центре аксиален (см. схему на с. 97). Поэтому из двух возможных аномеров моносахаридного остатка внутри полисахаридной цепи окислению подвергается только один. Если далее такой окисленный полисахарид подвергнуть мономерному анализу, то по исчезновению тех или иных моносахаридов из гидролизата (по сравнению с исходным полисахаридом) можно судить о том, что именно эти остатки в полисахаридной цепи имели «окисляемую» конфигурацию (с аксиальным водородом при С-1), а сохранившиеся – «неокисляемую» (с экваториальным водородом при С-1).
Описанный метод установления конфигурации гликозидных связей достаточно эффективен и широко применяется сейчас в структурных исследованиях за неимением лучшего. В нем, однако, заложено принципиальное несовершенство

Дело в том, что конформация моносахаридного звена в ацетилированной полисахаридной цепи обычно закреплена не жестко, так что даже при значительной предпочтительности одной конформации данного звена (например, с экваториальным водородом при С-1) оно в растворе может принимать, по крайней мере кратковременно, и противоположную конформацию (с аксиальным водородом при С-1). При этом «неокисляемое» звено, не меняя конфигурацию гликозидной связи, превращается в «оксиляемое» (см. схему). За счет этого строгая избирательность

реакции нарушается, и результаты такого анализа приходится трактовать не в четких терминах «да»-«нет», а в более осторожных:«преобладает»-«имеется в небольшом количестве». Более того, в определенных случаях анализ полисахаридов этим методом приводит и к прямо ошибочным заключениям:по видимому, вследствие значительных конформационных искажений звеньев за счет влияния всей полисахаридной цепи в целом.
Рассмотрим еще один путь исследования структуры полисахаридов, претераевающий в настоящее время бурное развитие и представляющийся исключительно перспективным:спектроскопию ЯМР на ядрах13С. Природный углерод состоит из трех изотопов:12С,13С и14С. Главный компонент12С и радиоактивный изотоп14С не дают сигнала в спектрах ЯМР, однако ядра13С, имеющие полуцелый
спин, дают такой сигнал. Содержание изотопа в природном углероде мало (1,1%). С одной стороны, это обстоятельство технически существенно осложняет спектроскопию на этих ядрах, а с другой – приводит к резкому упрощению картины спектров по сравнению со спектрами резонанса на протонах.
Дело в том, что в обычных органических молекулах ядра 12С и13С располагаются во всех возможных положениях по закону случая. Поэтому появление двух ядер13С в соседних положениях молекулы – событие маловероятное (вероятностью 0,0121%). В результате спин-спиновые взаимодействия13С-13С практически не оказывают влияния на картину спектров ЯМР13С, т.е. каждое такое ядро дает в спектре синглет (спин-спиновые взаимодействия с соседними протонами подавлены благодаря спекиальной технике съемки спектров).
Кроме того, величины химических сдвигов ядер 13С и различия между этими величинами велики по сравнению с величинами химических сдвигов протонов. Поэтому ЯМР-спектр на ядрах13С представляет собой набор достаточно хорошо разрешенных синглетов, что резко облегчает интерпретацию спектра. Величина химического сдвига определяется, как обычно, всем окружением данного ядра. В моносахаридах нет эквивалентных (в смысле химического окружения) положений углеродных атомов. В результате, например,13С ЯМР-спектр глюкозы состоит из шести хорошо разрешенных синглетов, отвечающих шести атомам углерода. Более того, спект равновесной смесиα-иβ-D-глюкопираноз (уравновешенная по мутаротацииD-глюкоза) представляет собой результат наложения двух независимых спектров аномеров, т.е. состоит из 12 сигналов.
При этом особенно важно, что присоединение гликозильного остатка к одному из атомов кислорода приводит к резкому (до 10 м.д.) изменению химического сдвига соответствующего ядра 13С, что позволяет определять положение межмономерных связей в полисахаридных цепях. Понятно, что основанный на такой спектроскопии метод обладает колоссальными возможностями для изучения полисахаридных структур. Разберем в качестве примера спектр агароподобного полисахарида одной из красных водорослей*.
Участок цепи, отражающий основные особенности структуры этого полисахарида, представлен на схеме, а его спектр – на рис.6.

В состав полисахарида входит четыре типа моносахаридных остатков (А-Г), различающихся по структуре или по окружению в цепи. Эти остатки содержат 25 химически различимых атомов углерода, в спектре же имеется 18 четко разрешенных сигналов. Таким образом, большинство углеродных атомов дает индивидуальные сигналы,
хотя меньшая часть из них – сигналы с перекрывающимися химическими сдвигами. Анализ спектра показывает (отнесения сигналов представлены на рисунке), что каждому из четырех моносахаридных звеньев соответствует, по крайней мере, один сигнал, свойственный углеродным атомам этого остатка, а углеродные ядра остатка А дают даже четыре сигнала, не перекрывающиеся ни с одним из сигналов других остатков и, следовательно, характерные только для него. Такой хорошо разрешенный спектр позволяет непосредственно установить природу моносахаридных остатков входящих в состав подобного полисахарида, положение межмономерных связей и конфигурацию гликозидных связей. Более того, по спектру можно измерить относительное содержание различных типов остатков в цепи.
Еще более интересно то, что химический сдвиг определенных ядер 13С в моносахаридных остатках зависит не только от структуры самого остатка, но и от структуры других звеньев, связанных с ним гликозидной связью. Так, остатки Б и Г (см. с. 99), взятые изолированно, представляются химически эквивалентными. Однако первый из них гликозилирован в положении 3 остатков 3,6-ангидро-L-галактозы (А), а второй – остатков 2-О-метил-3,6-ангидро-L-галактозы (В). В связи с этим химические сдвиги ядер С-3 остатков Б и Г оказываются различными (82,2 и 82,7 м.д. соответственно). Такая чувствительность положения сигналов в спектре13С ЯМР к окружению моносахаридного звена открывает захватывающие возможности для определения последовательности остатков в цепи.
Таким образом, спектроскопия ЯМР на ядрах 13С позволяет не только определять природу, тип связи, конфигурацию гликозидных остатков, входящих в состав биополимера, т.е. решать задачу мономерного анализа, но и устанавливать ближний порядок в расположении этих остатков в цепи, т.е. получать информацию, извлекаемую обычно из методов фрагментации. Принципиально важно, что такой анализ является неразрушающим. Поэтому весь полисахарид, использованный для съемки спектра, возвращается к исследователю в неизменном виде. В свете сказанного можно полагать, что в ближайшем будущем этот метод исследования станет одним из ведущих для изучения полисахаридных структур и заставит классические
Деструктивные методы значительно уступить свои позиции.
Мы рассмотрели несколько методов структурного анализа полисахаридов. Они далеко не исчерпывают всего арсенала инструментов исследования в этой области, но характеризуют многие важные принципы такого исследования. В смысле возможностей выяснения ближнего порядка моносахаридных звеньев в цепях и их дальнего порядка, а также оценки общего плана построения макромолекулы эти методы информативны далеко не в равной степени. Если, как мы видела, для характеристики ближнего порядка эти методы вполне пригодны, то с вопросами дальнего порядка дело обстоит гораздо менее благополучно. В сущности, все эти методы (заметим еще раз, основные методы структурного анализа полисахаридов) позволяют узнать о дальнем порядке очень мало, по крайней мере в общем случае. Это, однако, не эквивалентно бессилию современной науки перед проблемой установления структуры сложных полисахаридов. Есть несколько эффективных подходов к определению общей схемы построения цепей и дальнего порядка звеньев, хотя и носящих более частный характер, чем разобранные выше методы. Вот один из примеров.
В разветвленных полисахаридах довольно частый случай – расположение более лабильных к кислотному гидролизу звеньев в боковых цепях и более прочных – в главной цепи. Тогда при частичном гидролизе можно получить набор моносахаридов и низших олигосахаридов из боковых цепей и полимерный материал главной цепи. Такой результат позволяет сразу установить, каково в общих чертах распределение определенных типов остатков между главной и боковыми цепями, а также, что не менее важно, получить достаточные количества полисахарида упрощенной структуры (такие полимерные фрагменты часто называют деградированными полисахаридами) и установить его строение, т.е. решить уже более простую задачу, чем установление строения нативного (неизмененного) полисахарида.
Итак, мы видим уже два принципа установления структуры сложных полисахаридов:с одной стороны, путь от мономерного анализа, через расщепление на олигосахаридные блоки и реконструкцию последовательности моносахаридных остатков во все более длинных сегментах цепей, а с другой – путь от общего представления
о макромолекуле, как о целом, через последовательную локализацию целых групп или типов моносахаридных остатков в определенных крупных элементах структуры (в главной цепи, в боковых цепях, в узлах и т.д.) и до локализации отдельных моносахаридов в определенных положениях этих цепей. Есть еще и третий путь, в некоторых случаях дающий весьма обширную информацию о структуре. Он связан с применением ферментов, которые катализируют расщепление полисахаридных цепей.
Главная особенность ферментов, как инструментов структурного анализа полисахаридов – высокая, в некоторых случаях абсолютная, спекифичность их действия. Ферменты, расщепляющие полисахариды (полисахаридазы), как правило абсолютно специфичны к конфигурации гликозидной связи (например, фермент настроенный на гидролиз α-гликозидной связи, совершенно не действует наβ-гликозидные связи), абсолютно специфичны к размеру цикла моносахаридного остатка и высоко спекифичных к структуре и конфигурации самого моносахаридного звена. Кроме того, и это особенно важно для установления строения полисахаридов, полисахаридазы обычно высоко избирательны к типу связей и к структуре остатков в ближайшем окружении к расщепляемой гликозидной связи. Поэтому уже сам факт расщепления определенной связи данным ферментов нередко дает много сведений о ближнем порядке остатков в этом участке цепи (пример такой избирательности лизоцима мы уже рассматривали в другом аспекте).
Ферменты, расщепляющие полисахариды, бывают двух типов – эндоферменты и экзоферменты. Первые катализируют гидролиз некоторых гликозидных связей внутри полисахаридной цепи, т.е. расположенные достаточно далеко от обоих ее концов. Вторые способны вызывать гидролитическое отщепление только концевых моносахаридных остатков (в некоторых случаях концевых дисахаридных фрагментов). Не нужно думать, что процесс на этом останавливается:в полученном укороченном полисахариде есть свое концевое звено, которое в свою очередь подвергается атаке ферментом, если обладает нужной структурой и конфигурацией.
Как же можно использовать полисахариды в структурных исследованиях?Эндоферменты – это прежде всего реагенты для высоко избирательной фрагментации полисахаридных цепей;причем в отличие от кислотного гидролиза
такое избирательное расщепление позволяет делать заключение и о конфигурации разорванной гликозидной связи, и о природе самого гликозильного остатка, и нередко о его ближайшем окружении в цепи. Таким образом, это весьма эффективный метод и получения фрагментов, и определения ближнего порядка моносахаридных звеньев. Гидролиз разветвленных полисахаридов экзоферментами может служить тонким и специфичным методом получения деградированных полисахаридов путем избирательного отщепления боковых цепей. Для расщепления цепей, особенно в неразветвленных полисахаридах, экзоферменты иногда открывают уникальные возможности установления регулярности, т.е. дальнего порядка звеньев. Насколько мощным является данный инструмент, можно судить по следующему примеру.
Экзофермент, катализирующий гидролиз β-D-глюканов с 13-связями, в частности ламинарина, до глюкозы (избегая строгой номенклатуры, назовем его ламинаразой), высоко специфичен к типу гликозидных связей, т.е. расщепляет только связи 13. Поэтому, если изучаемый полисахарид под действием лиминаразы претерпевает полный гидролиз, можно уверенно утверждать, что он имеет регулярную структуру и построен только изβ-D-глюкопиранозных звеньев, соединенных13-связями. Иными словами, исследование полисахарида при помощи ферментативного гидролиза дает сразу сведения и о мономерном составе, включая конфигурацию и положение межмономерных связей, и о ближнем порядке звеньев, и о дальнем порядке остатков в цепях. На первый взгляд может показаться, что применительно к регулярному неразветвленному полисахариду ферментативный гидролиз дает информацию такого же характера, что и обычный мономерный анализ при помощи метилирования (если отвлечься от конфигурации гликозидных связей). Мы сейчас увидим, однако, что это не соответствует действительности.
Допустим, мы изучаем глюкан со степенью полимеризации (числом моносахаридных остатков в цепи) 100. В результате его анализа методом метилирования в нем были обнаружены только 13-связи и не обнаружено других. Однако «не обнаружено» – понятие довольно неопределенное. Его истинный смысл можно оценить, только зная чувствительность примененных аналитических методов. В нашем случае это чувствительность обнаружения минорных компонентов в смеси производных метилированных
моносахаридов. Для существующих сейчас методов анализа таких смесей оценка нижнего предела чувствительности в 1% очень хорошая. Это значит, что мы вполне могли не заметить одного «аномального» звена (например, 14-связи) на каждую полисахаридную молекулу. Таким образом, вывод о строгой регулярности строения на основании таких данных будет весьма шатким. Теперь посмотрим, что даст для такого полисахарида ферментативный гидролиз.
Экзоламинараза гидролизует такой полисахарид, последовательно отщепляя от него по одной молекуле глюкозы, начиная с невосстанавливающего конца. Если в цепи встретится одно «аномальное» звено (например, звено с 14-связью), то ферментативный гидролиз в этом месте прекратится и (это очень важно заметить) вся остальная часть полисахаридной молекулы останется нерасщепленной. Действительно, у такого «огрызка» цепи концевое звено аномальное и фермент перестает на него действовать, как показано на схеме этапов последовательного гидролиза полисахарида экзоферментом.

Г-свободная глюкоза или ее остаток, связанный «нормальной» для данного фермента связью;Г* - «аномальный»
остаток, глюкозидная связь которого не расщепляется ферментом.
Поскольку мы не знаем, в каком месте располагается аномальное звено, примем в первом приближении, что оно с равной вероятностью может оказаться в любом положении. Тогда при одном аномальном звене на молекулу (неопределенность метилирования) ферментативный гидролиз будет останавливаться в среднем в середине каждой цепи. Это означает, что выход образующейся глюкозы составит 50% от общего содержания в образце. Такое расхождение с результатами, ожидаемыми для регулярного полисахарида (100%), можно обнаружить даже самыми грубыми методами анализа. Реальная же точность определения глюкозы стандартными методами составляет несколько процентов. Скажем осторожно, 5%. Это значит, что, если при ферментативном гидролизе такого полисахарида мы получаем глюкозу с количественным выходом, то с учетом 5%-й погрешности мы вправе утверждать, что по крайней мере 90% всех полимерных молекул в образце имеют регулярное строение и не содержат аномальных звеньев. Таким образом, применительно к нашему примеру регулярность, установленная методом метилирования, на дает даже права утверждать, что в полисахариде есть молекулы, не содержащие ни одного аномального звена, а регулярность, установленная с использованием ферментативного гидролиза, позволяет сказать, что не менее 90% всех молекул образца регулярны в строгом смысле слова. Разница количественная, но явно переходящая в качественную.
Может показаться, что ферментативный гидролиз является идеальным методом исследования структуры полисахаридов, обеспечивающим исчерпывающую информацию обо всех уровнях организации макромолекулы – от подробной характеристики мономерного звена до структуры всей конструкции в целом. Это действительно так, но только для тех полисахаридов, для которых доступны (и, что не менее важно, хорошо изучены) наборы соответствующих ферментов. А это можно сказать далеко не обо всех типах полисахаридов. Чтобы понять, почему это так, надо совершить маленький экскурс в биологию.
Ферменты продуцируются живыми организмами. Организм синтезирует ферменты, расщепляющие определенный тип полисахарида, если встречается с этим полисахаридом в естественных условиях и если расщепление
такого полисахарида необходимо для его жизнедеятельности. Например, разветвленные глюканы типа амилопектина и гликогена почти универсально распространены в наземных растениях (амилопектин) и в животных организмах (гликоген) в качестве главного энергетического резерва. Для реализации этого резерва сами организмы нуждаются в соответствующих гидролитических ферментах. В не меньшей степени в таких ферментах нуждаются другие организмы – те, которые их поедают. Очевидно, например, что травоядные должны продуцировать ферменты, расщепляющие амилопектин (один из двух компонентов крахмала). Неудивительно поэтому, что такие ферменты можно выделить из многих доступных источников. Если же учесть большое значение, которое имеют такие ферменты для человека (и как ферменты собственно человеческого организма, и как важнейшие элементы биохимии сельскохозяйственных растений, и как активные участники многих процессов в пищевой промышленности и т.д.), то становится понятным, почему ферменты, расщепляющие гликоген и амилопектин, относятся к числу наиболее доступных и подробно изученных. Поэтому, в частности, именно в структурных исследованиях полисахаридов типа амилопектина и гликогена гидролитические ферменты являются сейчас основным инструментом, используя который удается почти исчерпывающе охарактеризовать их весьма сложные структуры.
Противоположным примером могут служить многие полисахариды водорослей, которые специфичны для этих растений и не встречаются в наземной флоре и фауне. Наивно было бы пытаться найти в наземных организмах расщепляющие их ферменты – этим организмам такие ферменты просто не нужны. Значит, источники подходящих полисахаридаз нужно искать в море, скорее всего среди травоядных морских животных. Вероятно, даже человеку, весьма далекому от биологии, понятно, что совсем не одно и то же – иметь источником фермента обычную лабораторную крысу или проростки картофеля, или пользоваться для этой цели, к примеру, морскими моллюсками, да еще не какими-нибудь вообще, а моллюсками определенного вида, обитающими в определенных районах и на определенных глубинах мирового океана. Чаще всего оказывается, что не только биохимия подходящего организма – продуцента нужного фермента – не изучена, но даже определить-то его как биологический
вид может лишь считанное число специалиситов во всем мире. И тем не менее использование ферментов является по-видимому, одним из основных и уж конечно наиболее плодотворных путей подробной характеристики структур полисахаридов. Только путь этот устлан далеко не розами*.
Так что же мы можем?
Мы рассказали о важнейших принципах и методах исследования структуры полисахаридов. Некоторые из них позволяют узнать о ближнем порядке моносахаридных остатков в цепи, другие дают сведения о дальнем порядке. Есть методы, пригодные лишь для изучения отдельных типов полисахаридов;есть и такие, область применения которых гораздо шире. К сожалению, среди них нет ни одного общего метода, который можно было бы использовать для изучения любого полисахарида (или хотя бы большинства из них), и нет ни одного метода, применение которого давало бы разумную степень уверенности в том, что с его помощью будет установлена полная структура полисахаридной молекулы. Нельзя даже сформулировать какое-то общее правило, следуя которому исследователь мог бы уверенно идти к цели в каждом новом случае:применяйте, например, такие-то и такие-то методы в такой-то последовательности, и вы сможете расшифровать полную структуру вашего полисахарида. Тем не менее строение множества полисахаридов известно сейчас с достаточной точностью и степенью подробности, причем нет оснований полагать, что эти структуры будут когда-либо опровергнуты. Возникает несколько парадоксальная ситуация:общих и надежных методов установления строения вроде бы нет, а структуры есть. Парадокс не устраняется каким-то простым способом, вопрос сложен и требует специального обсуждения.
Прежде всего, хотя известные методы структурного исследования действительно носят частный характер,
почти для каждого типа полисахаридов можно подобрать комбинацию приемов и методов, достаточно информативных для конкретного случая. Совокупность сведений, полученных различными независимыми путями позволяет охарактеризовать структуру уже достаточно полно:получить довольно подробные данные о ближнем порядке звеньев, об общем плане построения молекулы, о многих элментах дальнего порядка. В то же время даже применение всех мыслимых методов исследования обеспечивает сейчас лишь ограниченные возможности точной локализации всех моносахаридных остатков в цепях. Кроме того, любой известный метод структурного анализа дает максимальную информацию только для «удачных» с точки зрения его принципа систем. Так, например, агароза устроена исключительно выгодно для успешного применения частичного гидролиза, а глюкан овса на редкость «хорошо приспособлен» для анализа его структуры с помощью деградации по Смиту. Такие «выгодные» структры встречаются, разумеется, далеко не всегда или, говоря более точно, не для всякого типа структуры можно подобрать столь эффективный метод исследования. Существо возникающих трудностей не сводится, однако, к ограниченности методического арсенала или к техническому несовершенству имеющихся методов. Гораздо более принципиальный характер носят следующие обстоятельства.
Если не считать отдельных случаев гидролиза экзополисахаридазами, у нас пока нет возможностей перебрать полисахаридную цепь звено за звеном, выяснив тем самым полную и точную последовательность всех остатков. А деструктивные методы типа частичного гидролиза оставляют возможность для существования каких-то минорных невыявленных сегментов (как мы видели на примере агарозы). Поэтому структуры цепей, выведенные на основании даже очень подробного исследования, как правило, характеризуются некоторой неопределенностью, по крайней мере в отношении наличия (или отсутствия) какого-то числа отклонений, аномальных звеньев, а также в отношении ограниченной точности определения количественных параметров структуры (таких, например, как число разветвлений на макромолекулу). Расширение арсенала методов, примененных к данному полисахариду, и повышение их точности может, конечно, снизить верхнюю оценку для содержания миноров и для ошибки
в определении количественных параметров, однако не сможет устранить неопределенность полностью. В этом смысле процесс установления строения полисахарида и последующего уточнения его структуры есть асимптотическое приближение к недостижимой истине, тогда как надежно установленная ковалентная структура типичного низкомолекулярного соединения или такого биополимера, как белок, представляет собой истину «в последней инстанции», которая далее уже не может быть уточнена или поколеблена.
Другое обстоятельство, еще более фундаментального характера, позволяет поставить под сомнение целесообразность особо точного определения полисахаридных структур. Вспомним то, что говорилось о микрогетерогенности полисахаридных цепей. Благодаря этому явлению, в образце полисахарида, обычно содержится множество близко родственных структур. Поэтому локализация отдельных моносахаридных звеньев в цепях может быть достигнута с точностью, по крайней мере не большей, чем вариации структур молекул внутри образца, связанные с микрогетерогенностью. Принципиально можно, конечно, свести к минимуму структурные вариации такого типа, например, при помощи тех или иных химических или ферментативных обработок (вспомним, как был превращен нерегулярный полисахарид порфиран в производное регулярного полисахарида агарозы) и путем особо прецизионного фракционирования. Для таких полисахаридов со сведенным к минимуму разбросом структурных параметров можно, по крайней мере, в принципе, установить строение гораздо более точно.
Вопрос, однако, в том, что, собственно, будет отражать такая структура. Она не только не отвечает характеристикам биополимера в том виде, в каком он функционирует в живой клетке, но даже неспособна характеризовать каждый компонент смеси, составляющей реальный биополимер. Более того, значительные вариации структурных характеристик полисахаридных цепей существуют, по-видимому, и для различных представителей данного биологического вида, и для различных клеток одного многоклеточного организма, и для данного организма или данной клетки на разных стадиях развития или различных условиях существования. В этом смысле на углеводные биополимеры закон постоянства состава не распространяется.
Поэтому не исключено, что для понимания химических основ функционирования углеводсодержащих биополимеров в живых системах важны скорее несколько огрубленные, усредненные сведения о структуре, т.е. именно те, которые получаются при использовании современных методов исследования этих объектов. С другой стороны, для изучения микрогетерогенности как явления, понимания его биологического смысла и биосинтетических причин как раз важным кажется именно прецизионное, особо точное определение строения отдельных компонентов тех сложных смесей, какими являются такие биополимеры. Так что в столь сложном вопросе, как стратегия структурных исследований полисахаридов, оба, казалось бы взаимоисключающих, ответа на вопрос о целесообразной точности и глубине проникновения в материал оказываются правильными.
Так все-таки, способна или неспособна современная наука справляться со структурами сложных полисахаридов?Могут ли они быть установлены точно?В какой мере можно доверять структурам, фигурирующим в литературе в качестве известных?
Вопрос резонный, и обсуждается он вполне серьезно в профессиональных дискуссиях, хотя далеко не в столь прямолинейной форме. При этом высказываемые оценки расходятся довольно широко – от весьма оптимистических до весьма пессимистических. Авторы этой книги, хотя и имеют по этому вопросу определенное мнение, обязаны быть объективными и не навязывать свое мнение читателю. Поэтому мы попробуем изложить крайние точки зрения, акцентируя внимание на спорных моментах и не стремясь найти какой-то единственно правильный ответ. Вспомним поучительные слова лауреата Нобелевской премии профессора Роберта Робинсона:«Вообще-то, разница между наукой и религией заключается вот в чем:наука твердит, что, возможно, ошибается, тогда как религия утверждает, что наверняка права»*. Итак, суммируем основные доводы «за» и «против» возможности современной науки определять структуры полисахаридных цепей.
Аргументы «за». То, что говорилось о несовершенсте современных методов об ограниченности областей их применения, о трудностях определения дальнего порядка
в полисахаридных цепях, конечно, справедливо. Нельзя, однако, забывать о резком расширении возможностей наших методов при их совместном применении. Информация при этом не складывается арифметически, а скорее умножается. Вот, например, одна из таких схем. Частичный кислотный или ферментативный гидролиз удаляет боковые цепи. Из данных мономерного анализа и состава продуктов гидролиза можно рассчитать моносахаридный состав и длину боковых цепей. Остаток – главная цепь – устроен значительно проще, и для его анализа пригодны уже многие методы. Теперь тот же полисахарид подвергают деградации по Смиту, которая в благоприятном случае приводит к расщеплению главной цепи. Мы получаем, так сказать, две неполные карты структуры в разных красках. А их наложение друг на друга даст уже полную картину, потому что неопределенности различных путей не совпадают. И подобных комбинаций разных методов возможно множество, так что для большинства полисахаридов удается уже сейчас собрать почти исчерпывающую структурную информацию. Так что при достаточной настойчивости и изобретательности мы сможем расшифровать структуру любого полисахарида.
Возражение. Тем не менее, сколько бы мы ни уточняли структуру, всегда остается возможность где-то вставить один-два неучтенных моносахаридных звена, а то и целый сегмент, и такая структура будет приводить к тому же набору экспериментальных данных. Даже в самых простых случаях линейных регулярных полисахаридов можно предположить малого числа аномальных звеньев, которые остались незамеченными из-за ограниченной чувствительности аналитики. Так что в принципе мы вправе поставить под сомнение надежность почти всех известных полисахаридных структур, в том числе и тех, которые уже фигурируют в учебниках и считаются окончательными.
Контраргументы. Такой скепсиси носит слишком уж отвлеченный характер. Следует отталкиваться от живой практики. Достаточно посмотреть, как много сложнейших структур уже расшифровано, особенно в последние годы. Может быть, тот или иной конкретный вывод и удалось бы поколебать или даже опровергнуть. Но в целом картина вырисовывается логичная и внутренне непротиворечивая. По мере накопления данных полисахариды
удается все увереннее объединять в определенные классы с характерными типами структур, общие закономерности их композиции проглядываются все более отчетливо. А это подтверждает надежность каждого отдельного вывода. Характерный пример – О-антигенные цепи бактериальных липополисахаридов. Среди них многие ксеротипы имеют главную цепь регулярного строения, в которой повторяющимся звеном служит небольшой олигосахаридный фрагмент. И каждый новый представитель этого класса, структура которого становится известной, подтверждает это обобщение. А механизм биосинтеза этих полисахаридов, который был недавно установлен, позволяет понять, как такая регулярность возникает, и почему все эти полисахариды должны иметь именно регулярное строение. Так что структура каждого из этих биополимеров, взятая изолированно, возможно, и может быть поставлена под сомнение, однако в целом система знаний о данной группе полисахаридов оказывается весьма и весьма надежной и устойчивой.
Возражение:Не следует, однако, переоценивать значение таких обобщений. Иной раз трудно даже сказать, чего от них больше:пользы или вреда. Вспомним, как такие концепции появляются. Устанавливают строение нескольких первых, простейших представителей некоторого класса полисахаридов. Именно простейших (по той причине, что работа над ними приходит к концу раньше). Так как об этом классе еще ничего не известно, над структурами работают очень тщательно. И выясняется, что эти полисахариды регулярны (с теми, впрочем, оговорками, которые уже высказывались).
Рождается смелая гипотеза (именно гипотеза!) о том, что так же построены и другие полисахариды этого типа. Работа со следующими представителями идет уже легче:путеводной нитью служит гипотеза о регулярности;исследователи знают, что именно они должны найти:регулярную структуру. И, конечно, они ее находят. Даже подтверждают достаточно надежно, хотя иной раз уже не столь надежно, как для первых таких полисахаридов.
Дальше идет, что называется, «вал»: все новым и новым полисахаридам этого класса смелее и смелее (а значит, с меньшими основаниями) приписывают ожидаемый тип структуры. А потом мы бегло просматриваем литературу и видим, что структуры всех изученных полисахаридов этого класса прекрасно вписываются в общую
схему. И уверенно говорим о надежной системе знаний. А более внимательный взгляд покажет, что эта система базируется лишь на очень небольшом числе действительно подробно и максимально надежно охарактеризованных структур. И живет такая схема, кочует из книги в книгу, из обзора в обзор как нечто общепринятое, пока жизнь властно не заставит пересмотреть ее. И приходится заново перепроверять множество структур, а многое и опровергать.
Контраргумент. Но здесь уже есть преувеличение. Конечно, некоторая тенденция к переоценке таких удобных обобщений действительно существует. Но на самом деле выводы о типе структуры внутри класса строятся не на единичных примерах. Скорее уж ненадежные и сомнительные структуры единичны. Так что оснований для типизации полисахаридных структур бывает достаточно.
Возражение. Нельзя, однако, рассуждать по принципу:«если большинство, то и все». Математик сказал бы, что тут нет достаточных данных для полной индукции. Обобщать можно и нужно, но не следует забывать, что вывод о регулярности всех полисахаридов данного класса есть всего лишь гипотеза, а не закон природы. Так что будь то десятый или сотый представитель этого класса, к установлению его строения нужно относиться столь же ответственно и критично, как к установлению строения первого, и делать утверждение о регулярности можно лишь тогда, когда для этого есть прямые экспериментальные основания, а не аналогии с другими сходными полисахаридами.
Контраргумент. Но ведь так чаще всего и бывает. В конце концов, в большинстве работ авторы не утверждают ничего большего, чем им позволяет прямой эксперимент. И их выводы о структуре можно уточнить, но нельзя опровергнуть.
Возражение. Однако это не есть законченные четкие структуры. В любой из них найдется ряд усредненных, округленных, приближенных параметров. И о каждом, даже очень хорошо изученном, полисахариде остается еще узнать очень многое
Контраргумент. Тем не менее это максимум того, что можно сейчас сделать. И это совсем не мало. В конце концов, отражают наши структурные данные объективную реальность? Отражают! Позволяют на структурном, молекулярном, уровне показать и объяснить разницу между
различными полисахаридами? Позволяют! Дают основу для первых суждений о связи структуры и функции?В значительной мере дают. Так давайте же примем ту степень приближения, которая нам сейчас доступна.
На такой формуле, вероятно, и следует остановиться:полисахаридные структуры приближенны, и в этом надо отдавать себе ясный отчет;но это та максимальная степень приближения к истине, которая достигается современными методами. Методы же исследования полисахаридных структур необходимо еще совершенствовать и, что важнее, создать принципиально новые, которые позволили бы решить проблему миноров, подойти к вопросу о дальнем порядке звеньев в нерегулярных полисахаридах, словом, ответить на те вопросы, решение которых нам пока недоступно. Что же касается аргументов «за» и «против», то они не столько опровергают, сколько дополняют друг друга. Приведенную аргументацию нельзя отнести к тем или иным спорящим научным школам или отдельным личностям. Она отражает, скорее, разные грани одной области науки, или, еще вернее, разные состояния одного и того же Исследователя.