

Глава 4
Функция
Предварительные замечания
Еще сравнительно недавно в сознании биохимиков или молекулярных биологов углеводы как клеточные компоненты ассоциировались прежде всего и почти исключительно с энергетикой живых систем; так что на фоне фантастических свершений молекулярной биологии белков и нуклеиновых кислот роль углеводов в организме казалась многим если не третьестепенной, то во всяком случае сравнительно малоинтересной, почти тривиальной. Сейчас положение меняется довольно резко. Можно даже взять на себя смелость предположить, что современная наука находится на пороге «углеводного бума» – качественного скачка в области познания тонких и высоко специализированных функций углеводных систем в молекулярных механизмах весьма ответственных сторон жизнедеятельности.
Углеводы*присутствуют, причем в значительных количествах и в богатом «ассортименте», во всех живых организмах (за исключением типичных вирусов, если вирусы можно называть живыми организмами). Разнообразие их структуры поражает воображение, а огромные информационные возможности заставляют думать, что столь сложные молекулярные образования были «выращены» эволюцией для выполнения по меньшей мере столь же сложных и тонких функций.
В самом деле, экономия – один из самых фундаментальных и универсальных принципов живых систем. «… Представляется вполне вероятным, что биомолекулы, играющие роль строительных блоков, отбирались в процессе»
Эволюции по своей способности выполнять не одну, а несколько функций. Насколько нам известно, живые организмы в норме не содержат нефункционирующих соединений, хотя существуют биомолекулы, функции которых пока не ясны»*.
По мере углубления наших знаний о природе жизненных процессов вырисовывается картина сложной и многогранной роли углеводов в живых организмах. Среди известных сейчас функций углеводов мы находим и роль энергетического резерва, и роль главных структурирующих веществ, и роль эластиков, и роль смазки, и разнообразные информационные функции, и многое другое. Такую поразительную функциональность этого класса можно, по-видимому, понять на общих соображениях. Действительно, такие биологические монофункциональные биополимеры, как нуклеиновые кислоты, имеют один тип ковалентой структуры: это линейные одномерные цепи. Напротив, структуры высокомолекулярных углеводов представлены по крайней мере двумя молекулярными типами: линейными и разветвленными, не говоря уже о том, что среди разветвленных полисахаридов можно также выделить несколько крупных классов структур, и что организация поседовательностей мономеров в полисахаридных цепях может принадлежать к нескольким принципиально различным типам. Из такого разнообразия структур, естественно, следует и разнообразие функций.
Мы не будем здесь рассматривать сведения о биологической функции углеводов сколько-нибудь подробно:и потому, что эта область еще не сформировалась как стройная система взглядов, и потому, что, будучи химиками, мы не можем изложить эти вопросы вполне компетентно. Многие аспекты проблемы находятся сейчас в стадии интенсивных исследований, так что однозначное представление о них еще не сложилось в науке (хотя ниже и фигурируют некоторые дискуссионные трактовки, показавшиеся нам особенно интересными). Мы надеемся лишь помочь читателю убедиться в высокой специфичности углеводных структур и в исключительной важности их роли во всем механизме жизни, опираясь при этом на несколько характерных примеров. Такой подход лучше всего охарактеризовать словами Г. Мелвилла: “Я не собираюсь
приниматься за дело с научной систематичностью; с меня довольно будет, если я добьюсь желаемого результата перечислением отдельных фактов, известных мне… из личного опыта или от надежных людей:необходимые выводы, как я рассчитываю, вытекут сами по себе»*.
Энергия. Пища
В древнеиндийском эпосе – «Махабхарате» – читаем:«Нет положения более бедственного,- сказал Шамбара,- чем то, когда ни сегодня, ни завтра утром не предвидится еды»**. И в другом месте:«О царь царей, пища – это жизнь, все живое обязано своим существованием прежде всего пище. Что мешает смерти победить жизнь? Пища!»***. Как видим, уже философы древней Индии, с характерной для них склонностью мыслить глобальными категориями, понимали потребность в пище гораздо глубже, чем просто как врожденный инстинкт. В их глазах это общебиологический закон, указывающий на необходимость непрерывно поддерживать извне хрупкое образование – жизнь, потому что жизнь – не стационарное состояние, а процесс. Современная медицина измеряе эту поддерживающую функцию пищи в условных калориях, подчеркивая тем самым преимущественно энергетическую роль пищи (а не структурную, иначе мы, наверное, говорили бы не о «калорийности» рационов, а, скажем, об их «молярности»). В этой главе мы коротко рассмотрим некоторые химические аспекты обмена энергией в живых системах, первоисточником которой для животных организмов служит пища (в обычном понимании этого слова), а для фотосинтезирующих – лучистая энергия («энергетическая пища» в чистом виде).
Давно стало прописной истиной, что жизнь, в любой ее форме, есть непрерывная борьба против второго начала термодинамики, против всемирной тенденции к спонтанному повышению суммарной энтропии. Общеизвестно также, как именно жизни удается добиться победы в этой, казалось бы, безнадежной схватке: биосфера перехватывает
энергию солнечного излучения, запасает ее в форме энергии химических связей и затем расходует для обеспечения всей энергетики живых организмов и поддержания в них относительно низкого уровня энтропии до того, как эта энергия окончательно рассеется в виде тепла.
Углеводы стоят в начале и в конце этого грандиозного, непрерывно проходящего через биосферу потока энергии и энтропии: главными продуктами фотосинтеза являются гексозы, а главным источником энергии, удовлетворяющей повседневные потребности всех живых организмов, служитD-глюкоза.
Не будем разбирать ни реакции, протекающие при фотосинтезе, ни процессы, ведущие к утилизации энергии глюкозы: и те, и другие рассматриваются даже в современных школьных учебниках, не говоря уже об обширной научной литературе разной степени популярности. Однако с химических позиций небезынтересно задуматься о том, почему именно углеводам выпала ключевая роль во всей биоэнергетике.
Попытаемся мысленно сконструировать оптимальные структуры, пригодные для выполнения такой функции. В «техническое задание на проектирование» нам надо заложить два самых общих условия:материалом должны служить органические соединения*, а главной средой для планируемых процессов – вода. Для более поздних этапов эволюции нужно прибавить и третье условие – существование окислительной (кислородной) атмосферы. Проектируемая система – биосфера – должна функционировать в замкнутом пространстве планеты, практически исключающем обмен веществом с внешней средой; единственное, что поступает в систему извне,- это солнечная энергия.
Поэтому наша система, рассчитанная на длительное существование, по материальному балансу должна быть замкнута в цикл:исходные и конечные продукты ее функционирования должны быть идентичны. Это производство – производство живых организмов – сверхмноготоннажное. В нем позволительно использовать только самое дешевое и массовое сырье. Главное, что нам нужно – это основные элементы – органогены:углерод, водород, кислород и азот. Их наиболее распространенные источники на современной Земле – углекислый газ, вода и молекулярный азот.
Три названных вещества представляют собой наиболее устойчивые формы существования четырех элементов в нынешних геологических условиях. Иными словами, им соответствует минимум потенциальной энергии. Следовательно, любые органические соединения, содержащие эти элементы или некоторые из них, будут обладать определенным запасом энергии по сравнению с «основным состоянием» (CO2+H2O+N2) и будут способны выполнять нужную нам функцию. Задача, однако, поставлена на оптимизацию:какие типы соединений будут справляться с этой функцией наилучшим образом?
Будем и дальше рассуждать как химики. Органические соединения содержат углерод в более восстановленной форме, чем в CO2. В этом, в первую очередь, и заключается причина их энергоемкости в окислительной среде. Поэтому, оптимизируя решение по принципу наибольшей удельной емкости нашего аккумулятора, мы придем к наиболее восстановленным структурам – к предельным углеводородам. Вспомним, однако, что основная среда, согласно «техническому заданию»,- вода, в которой предельные углеводороды практически нерастворимы. Таким образом, и конструирование их молекул на стадии запасания энергии, и их распад на стадии утилизации энергии неизбежно включали бы гетерогенные реакции.
При прочих равных условиях гетерогенные реакции протекают резко замедленно по сравнению с гомогенными (это можно понять из самых общих соображений:гетерогенная реакция протекает на границе раздела фаз, т.е. в двухмерном пространстве, а гомогенная – в объеме, т.е. в трех измерениях). Поэтому биоэнергетика, построенная на предельных углеводородах, была бы в целом медленной: система была бы способна запасать много энергии, но обладала бы низкой мощностью. Это привело бы к большому замедлению всех жизненных процессов и, как следствие, к тому, что организмы оказались бы в гораздо большей степени подвержены воздействию неблагоприятных изменений внешней среды. Вывод:надо ориентироваться на более гидрофильные, растворимые в воде соединения, и при этом не слишком проиграть в энергоемкости по сравнению с углеводородами.
Повысить гидрофильность органической молекулы можно путем введения в нее полярных групп. Для создания таких групп пригодны два других имеющихся в нашем распоряжении элемента-органогена:кислород и азот.
При помощи каждого из них можно построить по три основных типа полярных группировок:карбоксильную (-COOH), карбонильную (=С=О) и спиртовую (С-OH) на основе кислорода, и амидиновую (-С(-NH2)=NH), имидную (=С=NH)и аминогруппу (С-NH2) на основе азота.
Что выбрать:кислород или азот?Предпочтение следует отдать кислороду. И вот по каким соображениям. Все три азотные группировки в водной среде в большей или меньшей степени склонны к гидролизу до соответствующих кислородных функций. Поэтому системы на их основе будут потенциально нестабильны, что нежелательно для столь фундаментальной системы, как биоэнергетика. Кроме того, все эти группы (особенно аминогруппа и тем более амидиновая) обладают основным характером. Поэтому рН растворов таких соединений будет сильно отклоняться в щелочную область от рН природной воды (около 7), что создаст для организмов с такой энергетикой дополнительные неудобства.
Из трех кислородных функций логично выбрать спиртовую, так как она соответствует наиболее низкой степени окисления углерода, а значит, наиболее высокому запасу энергии. Как показывает опыт органической химии, для того чтобы спирты хорошо растворялись в воде, идеально «вписывались» в ее структуру, нужно у каждого или почти у каждого углеродного атома иметь по одной гидроксильной группе. Так мы приходим к многоатомным спиртам типа HO-CH2-(CHOH)n-CH2-OH.
Создание таких молекул изCO2и воды (на стадии запасания энергии) и их расщепление (на стадии утилизации энергии) обязательно требуют образования и расщепленияC-C-связей. Оба эти процесса в алифатическом ряду обычно сопряжены с преодолением значительных потенциальных барьеров, что, разумеется, отрицательно сказывается на скоростях соответствующих реакций. Конечно, можно положиться на всемогущество биокатализаторов – ферментов, но все-таки лучше заранее облегчить им работу. Для этого надо подобрать какую-нибудь реакцию, которая легко протекает в водной среде и обратимо ведет к образованию С-С-связей в полиоксисоединениях.
Такая реакция есть:это альдольная конденсацияα-оксикарбонильных соединений и обратная ей реакция – ретроальдольный распад. Простейший ее пример – конденсация гликолевого альдегида по схеме:

Продуктами такой реакции являются полиоксиальдегиды. Для перехода к ним от многоатомных спиртов, к которым мы пришли на предыдущих этапах рассуждений, в структуру последних нужно внести лишь минимальные изменения: одну альдегидную группу. При этом мы проиграли в запасе энергии лишь минимально (один атом углерода оказался более окисленным, чем другие), но зато приобрели реакционную подвижность С-С-связей углеродного скелета, т.е. наиболее консервативного элемента структуры.
Теперь надо решить, какова должна быть оптимальная длина углеродной цепи в наших соединениях. Но здесь положение сущственно меняется. Вопрос о длине цепи природа материала решает за нас. Действительно, мы помним, что оксикарбонильные соединения легко и самопроизвольно замыкаются в кислородные гетероциклы, из которых наиболее устойчив шестичленный – пиранозный – цикл. При последовательном росте цепи полиоксиальдегида возможность замыкания пиранозного цикла впервые появляется у C5-оксиальдегидов, а если цепь наращивается сразу на два углеродных атома, как при конденсации гликолевого альдегида, то у соединенийC6. После замыкания пиранозного цикла карбонильная группа в молекуле исчезает (по крайней мере в явном виде) и дальнейший рост или распад цепи по схеме альдольной конденсации или ретроальдольного распада становится затруднительным. Так что подобные реакции автоматически останавливаются на молекулахC5 иC6, т.е. на моносахаридах альдопентозах и альдогексозах.
Теперь можно обратить внимание на то, что в полиоксиальдегидах все углеродные атомы, кроме концевых, являются асимметрическими центрами. Придавая им определенные
конфигурации, можно “нагружать” эти молекулы дополнительной информацией (т.е. “отрицательной энтропией”), что даст живым организмам добавочные преимущества в их противодействии второму началу термодинамики.
Пойдем дальше. Задумаемся над вопросом об оптимальной конфигурации этих центров. Из данных конформационного анализа легко заключить, что в ряду стереоизомерных полиоксиальдегидов наиболее устойчивым будет тот, у которого все заместители при пиранозном цикле экваториальны. Такая структура возникает в ряду альдопентоз для ксило-, а в ряду альдогексоз – для глюко-конфигурации. А универсальной единицей хранения энергии в реальных живых системах является D-глюкоза! Так, исходя из самых общих предпосылок и законов логики органической химии, приходим к тому же, к чему привела эволюцию стихийная логика природы.
Зададимся, наконец, вопросом, почему основным хранилищем энергии оказались углеводы, а не АТФ (аденозинтрифосфат), как известно, служащий единой «разменной монетой» в энергетике всех живых систем. Не лучше ли было бы и запасать, и хранить, и расходовать энергию в виде такого универсального вещества и не прибегать к помощи углеводов?Оказывается, что нет.
АТФ, как разменная монета удобна для покрытия мелких расходов и весьма мало пригодна для хранения сбережений. Действительно, при полном окислении глюкозы в клетке образуется 11 молекул АТФ (или его энергетических эквивалентов). Молекулярная масса глюкозы – 180, а натриевой формы АТФ – 573 дальтона. Таким образом, удельные энергоемкости глюкозы и АТФ относятся как 35:1. Так что глюкозы – гораздо более компактное хранилище энергии.
Существует способ еще несколько повысить удельню энергоемкость углеводов. Это поликонденсация моносахаридов в полисахариды. Вот по каким причинам организмы прибегают к этому способу.
Глюкоза чрезвычайно легко растворима в воде. Мы видим в этом важное преимущество моносахаридов как хранилищ энергии перед углеводородами. Но это преимущество, однако, не абсолютно и превращается в недостаток при необходимости депонирования значительных энергетических ресурсов. В самом деле, для этого в клетке должен был бы содержаться весьма концентрированный раствор глюкозы, что невыгодно и по физико-химическим,
и по биохимическим причинам. Поскольку глюкоза – низкомолекулярное соединение, это было бы связано со значительным повышением осмотического давления в клетке и с тенденцией легкой диффузии глюкозы наружу. Для противодействия подобным нежелательным факторам потребовалось бы создание сложных специализированных механизмов. С другой стороны, глюкоза представляет собой субстрат многих ферментативных реакций в клетке, и для ограничения ее чрезмерного расходования во время хранения клетка должна была бы располагать множественными и разнородными системами. Напротив, создание запасов энергии в форме специализированных полисахаридов (так называемые резервные полисахариды) обеспечивает живым организмам целый ряд преимуществ.
Поликонденсация моносахаридов в полисахариды сопряжена с отщеплением воды и, следовательно, со снижением молекулярной массы, отнесенной к потенциальной моносахаридной единице (для глюкозы – на 10%). Некоторая дополнительная энергия запасается в форме энергии образующихся гликозидных связей (гидролиз гликозидов – слабоэкзотермическая реакция). Резервные полисахариды высокомолекулярны, а большинство из них нерастворимо в воде при физиологических условиях. Так что все отрицательные эффекты хранения в клетках больших количеств свободной глюкозы снимаются.
Для утилизации энергии резервных полисахаридов путем их расщепления до моносахаридов нужно ограниченное число специфических ферментов – полисахаридаз. Это обеспечивает организмам возможность эффективного управления поступлением свободной глюкозы, т.е. в конечном счете расходованием запасенной энергии, как путем активации или угнетения этих ферментных систем, так и при помощи включения или блокирования биосинтеза соответствующих ферментов.
В животных организмах функцию резервного полисахарида выполняет гликоген, в большинстве растений – крахмал (амилоза+амилопектин), в бурых водорослях – ламинарин, в дрожжах и бактериях – декстраны. (Заметим, что все эти полисахариды построены только из остатков D-глюкопиранозы.) Высшие растения накапливают крахмал в особенно больших количествах в органах, связанных с воспроизведением вида, где необходимо создавать значительные энергетические ресурсы для обеспечения развития зародыша:в семенах при половом размножении
и корневищах – при вегетативном (их клетки часто оказываются буквально «битком набиты» крахмальными гранулами). Именно эти части растений составляю основу растительной пищи и главный источник пищевой энергии для современного человека (семена злаковых, клубни картофеля и т.п.). В организмах животных гликоген накапливается в первую очередь в клетках печени и мышц. Рассмотрим немного подробнее функцию гликогена печени.
В печени гликоген играет роль буфера глюкозы, циркулирующей в крови и являющейся главным энергетическим ресурсом всех клеток организма. Концентрация глюкозы в плазме крови должна поддерживаться постоянной: падение ее ниже нормы приводит к голоданию клеток и оказывается гибельным для тех из них, которые неспособны создавать собственные энергетические резервы (каковы, например, клетки головного мозга), а превышение ведет к резким биохимическим сдвигам в клетках, и также особенно опасно для клеток мозга. Между тем и расходование глюкозы плазмы, и ее поступление подвержены резким колебаниям. Например, при переходе от покоя к активной деятельности убыль глюкозы скачкообразно возрастает, а при переваривании пищи, особенно углеводной, в кровь быстро поступают значительные количества глюкозы. Таким образом, понятно, что организм должен располагать быстродействующими и легко управляемыми механизмами биосинтеза гликогена (депонирование избыточной глюкозы плазмы) и его расщепления (компенсация энергетических затрат). На примере расщепления гликогена удобно проследить связь его структуры с выполняемой функцией.
Гликоген построен из остатков α-D-глюкопиранозы и имеет высоко разветвленную структуру (см. с. 144). Связь остатков в цепях14, в точках разветвлений –16. Количественные параметры, характеризующие структуру гликогена варьируют, в зависимости от его источника (вида животного, природы ткани). В типичных случаях внешние неразветвленные цепи (А) содержат шесть-десять моносахаридных остатков, а во внутренних цепях (В и С) между разветвлениями находится два-четыре остатка глюкозы. Молекулярные массы гликогенов широко варьируюся и могут достигать десятков миллионов дальтон. (Это весьма значительная величина даже для биополимеров;она превышает, например, массу многих вирусных
частиц.) В отличие от большинства других резервных полисахаридов гликоген хорошо растворим в воде.


где I-общая схема структуры (А-концевые неразветвленные цепи, В-цепи, несущие разветвления, С-единственная в молекуле цепь с восстанавливающим концом,R-восстанавливающий конец);II-детальная структура участка, примыкающего к узлу разветвления;III-схема, иллюстрирующая первые две стадии ферментативного расщепления гликогена (кружки символизируют остаткиα-D-глюкопиранозы, горизонтальные стрелки –14-связи, вертикальные –16-связи).
Расщепление гликогена в печени катализируется двумя ферментами: гликоген-фосфорилазой иα-1,6-глюкозидазой. Оба фермента высоко специфичны к структуре отщепляемого остатка (отщепляют только концевой остатокα-D-глюкопиранозы), и к типу разрываемой связи (первый расщепляет только14-связи, второй – только16), и к структуре цепи, прилегающей к разрываемой связи со стороны восстанавливающего конца.
Особенно прихотлива специфичность последнего типа у фосфорилазы. Она отщепляет один за другим все 14-связанные остатки неразветвленных цепей (типа А) и оставляет неотщепленным последний остаток глюкозы, связанный с другой цепью16-связью в точке ветвления. В цепях такого же типа В расщепление происходит аналогично, но останавливается за несколько14-связанных остатков до точки разветвлкения. Далее на частично расщепленный гликоген действуетα-1,6-глюкозидаза, которая гидролизует16-связь одиночного глюкозного остатка, сохранившегося в точке ветвления после действия фосфорилазы. В результате остаток цепи типаBпревращается в цепь типа А, после чего включается следующий цикл реакций, катализируемой фосфорилазой, пока этот фермент вновь не «споткнется» на очередных разветвлениях, после чего вновь вступает в «игру» глюкозидаза и т.д.
Из описанной схемы ферментативного расщепления гликогена легко видеть, насколько целесообразно построена молекула гликогена в свете его основной функции. В самом деле, благодаря исключительно высокой разветвленности цепей гликогена каждая его молекула содержит большое число невосстанавливающих концов цепей (порядка десятков тысяч), так что одна молекула полисахарида может подвергаться атаке одновременно во многих местах. Это обстоятельство обеспечивает чрезвычайно высокую скорость расщепления и, следовательно, возможность почти мгновенной мобилизации заключенных в гликогене энергетических ресурсов.
Биологический смысл субстратной специфичности расщепляющих гликоген ферментов легко понять:«ключ» от энергетического «сейфа» – молекулы гликогена – должен идеально подходить к «замку», особенно если вспомнить, что это хранилище должно безотказно открываться в аварийных ситуациях (например, при стрессах). И в то же время, эти ферменты не должны действовать ни на какие другие углеводные структуры, иначе при их включении
клетки печени не только выдадут нужную организму глюкозу, но и быстро разрушат сами себя. Однако и замок должен точно соответствовать ключу. Иными словами (и тут мы вплотную подходим к вопросу о специфичности полисахаридных структур), вариации структуры гликогена допустимы лишь в таких пределах, в которых он сохраняет способность быть хорошим субстратом для расщепляющих его ферментов печени. А это, как мы видели, очень узкие пределы, не допускающие, например, ни наличия в молекуле фуранозных звеньев, ни изменения конфигурации гликозидных связей, ни наличия межмономерных связей, отличных о 14и16, ни замены части остатков глюкозы на остатки других моносахаридов и т.д. На любой из таких аномалий, даже если их содержание в молекуле незначительно, ферментативное расщепление гликогена приостановится, и вся остальная, и весьма значительная, часть полисахаридной молекулы не сможет быть вовлечена в обмен*.
Еще несколько слов о специфичности полисахаридов. Когда говорят о субстратной специфичности ферментов, обычно (особенно в научно-популярной литературе) делают акцент исключительно на способности фермента отличать свой субстрат от огромного числа других веществ, описывают фермент как изумительную распознающую машину, идеально приспособленную для выполнения своей функции.
При таком взгляде явно или неявно подразумевается пассивность субстрата в ферментативном акте, ему отводится роль инертного материала, над которым производится некая операция. Между тем современная концепция ферментативного катализа (концепция взаимно-индуцированного соответствия) отводит обоим компонентам взаимодействия фермент-субстрат равноправные активные роли. Суть ее в том, что при образовании фермент-субстратного комплекса происходит одновременное изменение конформации и субстрата, и фермента;это дает в итоге идеальную подгонку молекул обоих участников одна к другой.
Образно говоря, это не механическое наложение двух твердых предметов с комплементарной конфигурацией
(«ключа» и «замка»), а скорее, взаимная настройка гибких объектов. В каждом из участников такого взаимодействия, взятом изолированно, предсуществует не готовая матрица, соответствующая структуре другого участника, а лишь способность создавать такую матрицу под влиянием второго компонента.
Таким образом, мы можем уверенно говорить о высокой специфичности углеводов по отношению к катализирующим их реакции ферментам, об их способности узнавать «свои» ферменты среди множества других веществ с таким же правом, с каким мы говорим о субстратной специфичности этих ферментов*.
Опорные функции
Между животными клетками, с одной стороны, и растительными и бактериальными – с другой, имеется несколько кардинальных различий. К их числу относятся различия в среде обитания этих клеток. Клетки животного организма погружены в специально созданную жидкую среду – кровь или лимфу. Эти жидкости в известном смысле подобны по составу древнему Океану, в котором некогда возникла жизнь (часто говорят поэтому, что животные носят в себе частицу моря). Суммарные молярные концентрации низкомолекулярных веществ во внеклеточных жидкостях животного и в цитоплазме близки. Поэтому животные клетки находятся в осмотическом равновесии со средой, а их мембраны не подвергаются механическим нагрузкам за счет неравновесной диффузии воды внутрь клетки или из нее.
Растительные и бактериальные клетки находятся в совершенно другом положении. Внешней средой для них часто оказываются весьма разбавленные водные растворы – почти чистая вода, тогда как суммарная молярность содержимого клеток составляет величину порядка нескольких десятых. Свободная диффузия воды внутрь клетки, т.е. по градиенту концентрации, развивает в ней значительное избыточное давление (до 20 атм.), которое тонкая полужидкая мембрана выдержать не может. Поэтому такие клетки окружены жестким каркасом, называемым
клеточной стенкой, который принимает на себя осмотическое давление, придает клеткам определенную устойчивую форму и защищает от внешних механических воздействий. У многоклеточных растений именно прочность клеточных стенок обусловливает способность организма сохранять постоянную форму и противостоять гравитационным, ветровым, волновым и другим механическим нагрузкам.
У животных, лишенных внутреннего скелета (беспозвоночных), выработались те или иные приспособления, выполняющие опорные функции. В частности, у членистоногих, высокоорганизованного типа беспозвоночных, тело покрыто твердой внеклеточной оболочкой (кутикулой членистоногих), выполняющей функции наружного скелета: механической защиты организма и опоры для органов движения (общеизвестным примером могут служить панцири ракообразных). По наружному расположению и основной биологической роли кутикула у членистоногих может быть уподоблена клеточной стенке.
Молекулярную основу механической прочности и стенки бактериальной клетки, и кутикулы членистоногих составляют неразветвленные полисахариды, молекулы которых имеют конформацию жесткого стержня. Такая конформация характерна для полисахаридных цепей, в которых две связи элементарного звена (моносахаридного остатка) ориентированы в пространстве параллельно. Это возможно для пиранозных звеньев, соединенных 14-связями, если и гликозидный кислород, и кислород при С-4 связаны с циклом экваториально. Одна из наиболее типичных укладок таких звеньев в стержнеобразную макромолекулу, включающая антипараллельную ориентацию соседних остатков, показана ниже:
![]()
Основные типы полисахаридов, используемых живыми организмами для создания таких жестких стержневых структур, построены из14-связанных остатков моносахаридов сβ-D-глюко-кофигурацией. ЭтоD-глюкоза в целлюлозе (растительная стенка),N-ацетил-D-глюкозамин в хитине (кутикула членистоногих) иN-ацетил-D-глюкозамин+N-ацетилмурамовая
кислота в гликопептиде бактериальной стенки. При однотипности полисаъаридной основы эти механические каркасы указанных групп организмов резко отличаются по принципам организации надмолекулярной структуры и по этому признаку распадаются на три класса:стенка бактериальной клетки, стенка растительной клетки, кутикула членистоногих. Мы здесь коротко разберем структуру первых двух.
Смешанный биополимер, построенный из полисахаридных цепей и коротких пептидных фрагментов (высокомолекулярный гликопептид), составляет основу структуры стенки бактериальной клетки. Его полисахаридные цепи собраны из правильно чередующихся β-14-связанных остатковN-ацетил-D-глюкозамина (1) иN-ацетил-мурамовой кислоты (2). Последняя представляет собой простой эфирN-ацетил-D-глюкозамина (по положению 3) и молочной (L-α-оксипропионовой) кислоты.

Карбоксильные группы остатка мурамовой кислоты ацилируют N-конец коротких пептидов, создающих таким образом неуглеводные разветвления полисахаридной цепи 3. В состав этих пептидов входит лишь очень ограниченный набор аминокислот, в том числе совершенно необычные для живого мира аминокислотыD-ряда (D-аланин 4 иD-глютаминовая кислота 5). Обязательными компонентами таких пептидов являются двухосновная аминокислота (D-глютаминовая) и аминокислоты, содержащие две аминогруппы:L-лизин (6) или эритро-(7) иD-трео-α,-диаминопимелиновая кислота (8). Эта особенность позволяет пептидным цепям в свою очередь нести разветвления:-карбоксильная группа остатка глютаминовой кислоты ацилируетN-конец пентапептида, построенного из остатков глицина, а С-конец этого пентапептида ацилирует-аминогруппу остатка лизина или диаминопимелиновой кислоты (9).

Таким образом, пентаглициновые фрагменты играют роль поперечных связок, сшивающих полисахаридные и пептидные цепи в трехмерную сетку. Общая схема образующейся структуры представлена ниже:

Толстые линии на ней – это полисахаридные цепи, М и Г – остатки N-ацетилмурамовой кислоты иN-ацетил-D-глюкозамина соответственно;тонкие вертикальные линии – пептидные цепи (N-конец наверху), А1, А2и т.д. -
остатки аминокислот, волнистые линии – пентапептидный фрагмент из остатков глицина, латинскими буквами обозначены его N-иC-концы.
Весь этот гликопептид стенки представляет собой одну гигантскую макромолекулу в форме мешка, имеющую молекулярную массу порядка десятков миллиардов дальтон. Высокая механическая прочность этой системы обусловлена сшитой структурой и жесткостью стрержнеобразных полисахаридных цепей, составляющих каркас всей конструкции. Помимо своей основной механической функции, гликопептид клеточной стенки принимает участие и в ряде других важных биологических феноменов. Упомянем кратко некоторые из них.
Биосинтез гликопептида стенки проходит через несколько этапов, включающих образование полисахаридных цепей, наращивание на них пептидных разветвлений и в заключение – сшивание этих пептидов пентаглициновыми мостиками. Ряд антибиотиков блокирует определенные стадии этого процесса, что в итоге приводит к нарушению биосинтеза стенки и, следовательно, к появлению нежизнеспособных бактериальных клеток после деления. Так, бацетрацин и ванкомицин ингибируют биосинтез полисахаридных цепей гликопептида, а пенициллин угнетает заключительный этап – образование пентаглициновых сшивок. Гликопептид рассматриваемого типа – общая основа клеточной стенки самых разнообразных бактерий;в то же время подобные структуры отсутствуют в клетках животных организмов. Отсюда становятся понятными причины широты антибактериального спектра таких антибиотиков, с одной стороны, и их исключительно низкая токсичность для животных, с другой.
Полисахаридные цепи гликопротеида стенки химически весьма устойчивы. Тем не менее, их гидролиз легко протекает под действием специфического фермента – лизоцима, весьма распространенного в живых организмах. Обработка многих бактерий лизоцимом приводит к разрушению стенки и в обычных условиях к гибели бактериальной клетки (из-за способности лизировать, т.е. растворять бактериальные клетки, фермент и получил свое название). Ряд слизистых выделений животных организмов, таких, как слезы или слюна, содержит лизоцим, что обусловливает их защитный эффект против вторжения инфекции.
Стенка растительной клетки, в отличие от гликопротеинового каркаса бактериальной, построена на иных принципах

структурной организации. Растительная стенка – надмолекулярная структура, отдельные компоненты которой не связаны ковалентными связями, а держатся вместе за счет межмолекулярных взаимодействий, в которых доминируют водородные связи. По композиции эта структура может быть уподоблена армированным материалам типа железобетона или стеклопластика, а по сложной многослойной конструкции более всего напоминает автомобильную покрышку (если искать аналогии среди знакомых макроскопических объектов). Механическую основу стенки (ее корд) составляют микрофибриллы целлюлозы – пучки тесно связанных линейных макромолекул, а цементирующим материалом служат полисахариды других классов – гемицеллюлозы и пектиновые вещества, а также аморфный ароматический полимер – лигнин.
Типичная стенка растительной клетки (рис. 7) состоит из нескольких слоев. В ней различают первичную стенку, составляющую наружный слой клетки, и вторичную стенку*, состоящую из внутреннего, среднего и внешнего слоев. В первичной стенке микрофибриллы не имеют определенной ориентации и переплетены в беспорядочную сеть. Во внешнем слое вторичной стенки они образуют два семейства параллельных линий, пересекающихся почти под прямым углом и образующих, таким образом, правильную («декартову») сетку. В среднем слое вторичной стенки микрофибриллы параллельны друг другу и почти параллельны оси цилиндра (фигуры, в грубом приближении

описывающей геометрию типичной растительной клетки). Наконец, во внутреннем слое вторичной стенки микрофибриллы также параллельны, но ориентированы под углом к оси. Такая сложная композиция обеспечивает исключительно высокие прочностные характеристики стенки растительной клетки (а следовательно, и растительных материалов, применяемых человеком).
Общая схема строения микрофибрилл в настоящее время выяснена довольно полно (главным образом с помощью электронной микроскопии и рентгеноструктурного анализа), хотя целый ряд подробностей еще продолжает дискутироваться. Микрофибриллы представляют собой агрегаты из нескольких так называемых элементарных фибрилл, в которых молекулы целлюлозы вытянуты продольно, а в поперечном направлении плотно упакованы в высоко упорядоченную кристаллическую структуру. Элементарная фибрилла (рис. 8) представлена стержнем с почти квадратным сечением (угол при вершине 86,5) и стороной 35Ǻ. На сечение приходится 36 цепей целлюлозы*. В поперечном сечении элементарной фибриллы молекулы целлюлозы упакованы в правильную решетку и соединены между собой водородными связями. Соседние молекулы ориентированы антипараллельно, т.е. направление гликозидных связей у них противоположно. Примыкающие одна к другой антипараллельные цепи целлюлозы организованы в пары, между которыми образуются особенно прочные водородные связи. У концов отдельных молекул возникают дислокации, в которых соседние молекулы претерпевают небольшой изгиб, после
чего их продолжения вновь встраиваются в кристаллическую решетку, обеспечивая тем самым целостность и общую упорядоченность элементарной фибриллы.
Внутри кристаллических областей элементарной фибриллы молекулы целлюлозы имеют строго упорядоченную конформацию жесткого стержня, в котором соседние глюкопиранозные остатки повернуты один относительно другого на 180. Такая конформация закреплена внутримолекулярными водородными связями, соединяющими каждую пару соседних остатков. Гидроксильная группа при С-3 выступает в роли донора протона, а кислород соседнего слева остатка – в роли акцептора. Фрагмент такой структуры представлен ниже (10):

Помимо строго упорядоченных кристаллических участков, структура которых была только что рассмотрена, в нативных микрофибриллах имеются также и аморфные (или, по крайней мере, менее упорядоченные) участки, где целлюлозные цепи упакованы более рыхло. Вопрос об относительном содержании аморфных и кристаллических участков и об их взаимном расположении в элементарных фибриллах и микрофибриллах до сих пор остается дискуссионным из-за отсутствия бесспорных методов для прямого экспериментального определения этих характеристик. Однако факт существования в нативных целлюлозах двух типов надмолекулярных структур не вызывает сомнения. Такие участки резко различаются по реакционной способности, что непосредственно проявляется в химических экспериментах.
Действительно, в кристаллических участках молекулы целлюлозы упакованы плотно и с большой энергией межмолекулярной связи. Поэтому молекулы, находящиеся внутри такого участка, недоступны для реагентов и могут вступать в какие бы то ни было реакции лишь после разрушения или удаления молекул, лежащих на поверхности и вступающих в прямой контакт с реакционной средой. В этом смысле кристаллические участки ведут себя в гетерогенных реакциях как классическое твердое тело. В аморфных участках отрезки молекул упакованы менее
плотно и более подвижны относительно друг друга. Внутрь этих участков может проникать и растворитель, и реагент;может происходить набухание. В результате образуются локальные гелевые структуры с большим содержанием жидкой фазы, соприкасающейся со всеми или с большинством молекул целлюлозы. Поэтому реакции полисахарида в таких участках по кинетическим характеристикам приближаются к реакциям в растворе, т.е. могут протекать значительно быстрее, чем истинно гетерогенные реакции в кристаллических участках. Так, например, при осторожном кислотном гидролизе нативной целлюлозы разрыв гликозидных связей происходит в первую очередь и почти исключительно в аморфных участках. Таким путем можно, в частности, получить препаративно высоко кристаллическую целлюлозу, состоящую почти исключительно из кристаллических «обрезков» и непосредственно измерить среднюю длину кристаллических участков. По разным определениям (и в зависимости от источника целлюлозы) длина кристаллических участков, измеренная подобным образом, колеблется о 500 до 3300Ǻ.
Помимо чисто научного интереса, который естественно вызывает структура такого уникального образования, как стенка растительной клетки, вопрос этот имеет крупное практическое значение. Знание тонкой структуры и подробностей формирования микрофибрилл и клеточной стенки в целом составляет солидную часть научного фондамента целлюлозной промышленности и производства натурального и искусственного волокна на основе целлюлозы. Характерным примером может служить непосредственная связь гелеобразующих свойств таких синтетических производных целлюлозы, как карбоксиметилцеллюлозы и частично метилированные целлюлозы, с распределением аморфных и кристаллических участков в исходном целлюлозном материале.
Маркировка поверхностей
Есть хорошо известный эксперимент: если губку, примитивное многоклеточное животное, расчленить до отдельных клеток, а затем оставить суспензию этих клеток в покое, то через некоторое время они вновь объединятся в многоклеточный организм. Если смешать суспензии клеток из губок двух различных видов, то при их объединении самопроизвольно
возникнут два агрегата, каждый из которых состоит из клеток одного вида. Сходным образом ведут себя в культуре эмбриональные клетки высших животных:они объединяются порознь по тканевому признаку. Вопрос о том, каким образом клетки «узнают» друг друга, чрезвычайно важен не только для понимания их поведения в искусственных условиях подобных экспериментов. Взаимное опознание клеток играеи решающую роль уже на самых ранних стадиях развития зародыша многоклеточного организма, когда начинается формирование определенных типов тканей, а затем органов. Близкородственные аспекты межклеточных взаимодействий имеют непосредственное отношение к проблеме злокачественных опухолей, тканевой несовместимости, иммунитета и многим другим биологическим феноменам.
Если животные клетки в подходящей искусственной среде поместить на твердую поверхность (например, на дно чашки Петри), то их деление будет происходить упорядоченно:на поверхности растет одноклеточный слой, а после того, как вся она будет покрыта клетками, деление практически прекращается – наступает так называемое контактное торможение. В этом эксперименте проявляются в сильно упрощенном виде те явления, которые определяют постоянство размеров и формы органов и всего взрослого многоклеточного организма. По-иному ведут себя в таких экспериментах раковые клетки:они образуют бесформенную клеточную массу, их деление не приостанавливается после заполнения поверхности одноклеточным слоем. В отсутствии такого торможения заключена главная причина злокачественности – бесконтрольного роста опухоли. Целостность нормального органа поддерживается прочными межклеточными связями. В опухолях эти связи значительно слабее:отдельные их клетки легко отделяются от основной массы, уходят в кровяное русло и разносятся по всему телу. В этом первопричина метастазирования – второй грозной особенности злокачественных опухолей.
Очевидно, что во всех явлениях подобного типа соприкасающиеся поверхности клеток передают некоторый сигнал – команду на связывание клеток, на торможение деления и т.п. Долгое время о природе веществ, ответственных за такие межклеточные взаимодействия, а также за многие классы специфических взаимодействий типа клетка-макромолекула, ничего не было известно. Позднее
стали накапливаться данные в пользу того, что во многих таких явлениях ключевая роль принадлежит углевода, находящимся на периферии клеток и макромолекул. Наиболее старым и хорошо изученным примером могут служить антигенные полисахариды многих бактерий. Например, капсула пневмококков, к числу которых относятся возбудители такого инфекционного заболевания, как пневмония, содержит значительные количества специфических полисахаридов. Именно на них вырабатывается иммунный ответ организма-хозяина. Доминирующая роль полисахаридов как антигенов таких бактерий доказана не только лабораторными, но и клиническими экспериментами. Так, еще во время второй мировой войны в американской армии была успешно осуществлена иммунизация большой группы людей против пневмонии путем введения им препаратов полисахаридов из пневмонийных бактерий.
Иммунная реакция – яркий пример взаимодействия типа клетка-макромолекула. По наиболее принятым сейчас представлениям упрощенная схема выработки иммунного ответа на бактериальный антиген такова. В лимфе и крови циркулируют специализированные клетки:Т- и В-лимфоциты, на поверхности которых находятся рецепторы к потенциальным антигенам. У каждого лимфоцита (точнее, у каждого клона, т.е. у семейства генетически тождественных лимфоцитов) имеются свои, индивидуальные для него рецепторы к потенциальным антигенам.
Разнообразие этих рецепторов (и клонов лимфоцитов) огромно:число различных рецепторов составляет величину порядка миллиона, так что практически на любой чужеродный биополимер (антиген) находится соответствующий ему рецептор. Зрелые В-лимфоциты, не соприкасавшиеся со «своими» антигенами (их называют «девственными» лимфоцитами), не делятся. Однако контакт с антигеном, например с бактериальным полисахаридом, служит сигналом для целой цепи событий. В-лимфоцит после этого трансформируется в плазматическую клетку и начинает делиться. Общее количество клеток данного клона резко возрастает;они начинают продуцировать и секретировать в кровь и лимфу большие количества свойственных этому клону иммуноглобулинов, т.е. антител, специфичных к данному антигену. Антитела реагируют с соответствующими антигенами в растворе, что приводит к их осаждению, и с теми же антигенами на поверхности бактериальной клетки. Таким образом происходят удаление
бактериальных токсинов (антигенов) из организма и атака на клетки возбудителя, в чем и заключается защитный смысл иммунного ответа.
Механизм распознавания рецепторами и антителами своих антигенов почти неизвестен. Известно, однако, что это в высшей степени специфичное взаимодействие, так как его блокируют самые незначительные изменения в структуре углеводного антигена. Структурно измененный полисахарид или углеводсодержащий биополимер – это уже другой антиген;на него реагируют рецепторы других лимфоцитов и вырабатываются другие антитела.
При выработке иммунного ответа клеточные рецепторы реагируют на углеводные детерминанты макромолекулы антигена. Обратным примером может служить взаимодействие клеток с макромолекулами холерного токсина. Последний представляет собой белок, в состав которого входят две высокомолекулярные пептидные субъединицы. Одна из них ответственна за первичное взаимодействие с клетками организма-хозяина, а другая – за токсический эффект. Было установлено, что рецептором на поверхности клеток, осуществляющим узнавание молекулы токсина и связывание с ним, является гликолипид – ганглиозид GМ1, в молекуле которого к липидной части присоединен олигосахаридный фрагмент, содержащий остаток сиаловой кислоты. После присоединения токсина к ганглиозиду от первого отщепляется токсическая субъединица, под действием чего происходит ряд изменений в активности ферментов клетки, в первую очередь активация аденилат-циклазы, а это в конечном итоге приводит к крупным нарушениям клеточного метаболизма и гибели клетки.
Во многих случаях структура углеводных цепей, находящихся на поверхности клетки или макромолекулы, служит своеобразной маркировкой, кодирующей адрес, по которому они должны быть доставлены при транспортировке в организме. Так, например, в мембране эритроцитов имеются гликопротеины, на наружных концах углеводных цепей которых находятся остатки нейраминовой кислоты. При наличии этих остатков эритроцит достаточно долго циркулирует в кровяном русле, а при их удалении быстро уходит из него. Полагают поэтому, что концы цепей, обнажающихся при удалении остатков нейраминовой кислоты, служат участками связывания эритроцитов с мембранами кроветворных и выделительных органов.
«В связи с этим возникает предположение, согласно
которому присоединение нейраминовой кислоты к гликопротеинам клеточной поверхности происходит в месте образования эритроцитов, например в костном мозге. Можно думать, что присоединение нейраминовой кислоты каким-то образом экранирует якорные участки эритроцита, т.е. реализуется команда «отдать концы» и корабль-эритроцит уходит в плаванье в кровоток»*.
Имеется ряд данных, указывающих на то, что в процессах клеточной дифференциации, т.е. при формировании тканей и органов в эмбриональной стадии, при специфической агрегации клеток (как в опытах с губками) большую информационную роль играют сульфатированные мукополисахариды – сложные биополимеры, в состав которых входят большой пептидный фрагмент и длинная полисахаридная цепь, включающая остатки уроновых кислот, аминосахаров и сульфата. Состав и структура этих соединений весьма индивидуальны не только для различных биологических видов, но и для различных тканей одного и того же организма.
Каков же все-таки механизм распознавания углеводных маркеров рецепторами поверхности клеток?В последние годы появилось одно чрезвычайно интересное объяснение такого феномена для ряда типов межклеточных взаимодействий. Чтобы его изложить, нужно напомнить некоторые положения энзимологии.
Ферменты – высоко специфичные катализаторы. Их специфичность двоякая. Во-первых, это субстратная специфичность, означающая, что фермент катализирует реакцию одного определенного вещества – субстрата, или по крайней мере субстратов, имеющих определенный общий элемент структуры. Во-вторых, это специфичность по реакции, суть которой в том, что из нескольких, а часто из многих реакций, в которые субстрат мог бы вступать в данных условиях, фермент катализирует только одну.
В активном центре фермента, т.е. в том месте белковой глобулы, в котором непосредственно осуществляется каталитический акт, имеется два участка с различными функциями. Участок связывания осуществляет соединение субстрата с ферментом и ориентацию его молекулы, оптимальную для осуществления основной реакции, но не участвует в реакции как таковой. Этот участок точно
«настроен» на структуру субстрата и «ответствен» за субстратную специфичность. Природа связи между субсратом и участком связывания может быть очень различной;но для нас сейчас важно уяснить, что эта связь достаточно прочная. Продукт такого взаимодействия называется фермент-субстратным комплексом.
Второй участок в активном центре – каталитический. Его задача – осуществлять реакцию субстрата, ориентированного и подготовленного к этому в составе фермент-субстратного комплекса. После реакции субстрат в составе комплекса превращается в продукт, т.е. образуется фермент-продуктный комплекс, который легко диссоциирует. Продукт уходит в среду, а фермент регенерируется.
В обычных условиях ферментативной реакции фермент-субстратный комплекс – образование эфемерное, так как сразу же после его возникновения происходит основная реакция. Однако, если реакция по тем или иным причинам заторможена (например, при низкой температуре), то фермент-субстратный комплекс становится способным к длительному существованию. Такая ситуация возникает, в частности, для ферментативной реакции типа A+BC+Dпри недостатке одного из субстратов. Например, фермент и субстрат А образуют нормальный комплекс, но в отсутствие субстрата В он не способен к дальнейшему превращению, и потому стабилен. Именно такой механизм образования стабильного фермент-субстратного комплекса, согласно излагаемой гипотезе, лежит в основе специфического, взаимного распознавания и сцепления клеток в ряде случаев межклеточных взаимодействий.
На поверхности таких клеток – на внешней стороне их мембран – находятся недостроенные углеводные цепи и ферменты (так называемые гликозил-трансферазы), переносящие на эту цепь недостающий моносахаридный остаток с предшественника – нуклеозид-дифосфат-сахара (НДФС), играющего при таком переносе роль второго субстрата. Однако в пределах одной клетки гликозил-трансферазы и их субстраты (недостроенные углеводные цепи) непосредственно взаимодействовать не могут, так как на мембране они пространственно разделены. Зато такое взаимодействие легко и эффективно осуществляется при контакте двух таких клеток: гликозил-трансфераза одной клетки образует фермент-субстратный комплекс с углеводной цепью другой клетки, и наоборот (рис. 9).

В отсутствие донора гликозильного остатка (НДФС) фермент-субстратные комплексы стабильны и связи между клетками прочны. При появлении же НДФС происходит достройка цепи и диссоциация комплекса, а следовательно, и связи между клетками. Такой механизм может превосходно объяснить высокую специфичность распознавания клетками друг друга и их ассоциирования, а также указывает путь тонкого регулирования взаимодействия: в отсутствие НДФС клетки ассоциируют, а при поступлении НДФС в межклеточное пространство диссоциируют. Такой механизм взаимодействия клеток позволяет понять, почему самые минимальные изменения в структуре углеводных цепей поверхности клетки решающим образом сказываются на ее взаимодействии с другими клетками или макромолекулами и на судьбе в организме (как в приводившемся выше примере с эритроцитами).
В настоящее время участие подобного механизма распознавания постулируют с различными степенями уверенности для целого ряда важнейших биологических явлений: формирования тканей и органов на эмбриональной стадии развития, взаимного опознавания некоторых половых клеток при оплодотворении, первичного акта тромбообразования – адгезии тромбоцитов на коллагеновых нитях
(связывание гликозил-трансфераз тромбоцита с углеводными цепями коллагена), объяснения различий поведения нормальных и опухолевых клеток и др. Было, например, показано, что опухолевые клетки обладают повышенной способностью к переносу гликозильных остатков на концы цепей той же клетки, на которой находится данная гликозил-трансфераза. Отсюда – пониженная способность к ассоциированию и подавление контактного торможения.
Можно полагать, что описанные представления окажутся чрезвычайно плодотворными для решения многих общебиологических проблем, а применительно к углеводам послужат толчком для гораздо более глубокого изучения их информационных, сигнальных функций в живых организмах на молекулярном уровне.
Молекулярная биология
полисахаридов
Огромные успехи исследований механизмов кодирования наследственной информации и биосинтеза белка, ферментативного катализа и регулирования активности ферменов, действия антибиотиков и гормонов, всей той области изучения живого, которую принято называть молекулярной биологией, приучили всех к мысли о том, что в структурах молекул жизни положение буквально каждого атома строго обусловлено и подчинено выполнению предназначенных для этих молекул биологических функций. Именно в этом смысле принято обычно говорить о специфичности биополимеров, прочно ассоциировавшейся в сознании исследователей с однозначным соответствием между структурой и выполняемой функцией. При таком «комплексе структурного детерминизма» трудно было освоиться с представлением о специфичности полисахаридов, для многих из которых характерна статистичность структур, микрогетерогенность и, нередко, хаотичность распределения моносахаридных остатков по цепи. И, тем не менее, накапливающийся материал по сложному и высоко специализированному функционированию углеводных полимеров в живых системах убеждает в том, что и в этой области возможен и необходим перевод функциональных свойств биополимеров на язык молекулярных структур, т.е применим основной принцип молекулярной
биологии. Только оперировать при этом нередко приходится с иными уровнями специфической упорядоченности их структур. Носителем функциональной специфичности, если можно так выразиться, здесь оказывается не некоторая одна ковалентная структура, а определенный тип ковалентной структуры. И этот тип может быть не менее строго детерминирован, чем единичная структура. Поэтому возникает задача сформулировать те типовые (а не индивидуальные) характеристики популяции различных молекул, составляющих углеводный биополимер, которые ответственны за его биологические функции. Именно такой подход к молекулярной биологии полисахаридов помог получить первые реальные успехи в этой области. Они связаны в первую очередь с именем Д.А.Риса (Англия), на идеях и результатах исследований которого в основном построено наше дальнейшее изложение.
Характерное свойство многих классов полисахаридов есть способность к гелеобразованию в водных растворах. Именно с этим свойством связан ряд биологических функций полисахаридов (а также ряд областей практического применения самих полисахаридов и их производных). Сюда, в первую очередь, относится обеспечение нужного набора механических свойств опорных систем (таких, например, как клеточные стенки), склеивающих и пластических свойств межклеточного вещества, упругости ряда систем (хрусталик глаза), функционирования смазочных материалов в живых организмах (синовиальная жидкость в суставах), материала поверхности эпителиальных клеток, вдоль которых движутся биологические жидкости (кровь, лимфа и т.п.), и других физико-механических и физико-химических характеристик строительных материалов живых систем. Очень наглядно роль гелеобразующей способности полисахаридов в обеспечении важных биологических функций можно проследить на следующем примере.
Водоросли прибрежных зон океана обитают в весьма своеобразных условиях:они подвергаются значительным волновым нагрузкам, направление и величина которых меняется по всем трем координатам в зависимости от ветров и микрорельефа, а водоросли литорали подвержены, кроме того, периодическим обсыханиям во время отливов. Чтобы противостоять таким воздействиям, красные и бурые водоросли выработали чисто «полисахаридное» приспособление. Все они содержа в качестве межклеточного вещества специфические кислые полисахариды (альгиновые
кислоты в бурых водорослях и сульфатированные галактаны в красных), способные даже в разбавленных растворах образовывать прочные упругие гели, удерживающие значительные количества воды в «псевдотвердом» состоянии.
Именно упругий гель оказался той механической основой, которая идеально приспособлена к сопротивлению неподвижных организмов волнам, и одновременно великолепным аккумулятором воды, позволяющим водорослям литорали благополучно переживать отливы. Не случайно, именно бурые и красные водоросли служат важным источником гелеобразователей, применяемых в промышленности. Гелеобразующая способность относится к тем немногим функциональным свойствам углеводных биополимеров, которые в настоящее время удается трактовать на молекулярном уровне.
Для образования полисахаридного геля нужно, чтобы цепные молекулы были организованы в рыхлую пространственную сетку, в ячейках которой находится растворитель (вода). Одним из ключевых вопросов, ответ на который позволяет связать структуру полимера с его способностью к гелеобразованию, является природа узлов этой сетки. Это могут быть ковалентные связи между цепями, и в таком случае сетка представляет собой одну гигантскую трехмерную молекулу. Так построен, например, гликопептид бактериальной стенки, который мы уже рассмотрели, а из искусственных образований – сефадекс, полусинтетический материал для гель-хроматографии. Более типичны полисахаридные гели, в которых связи цепей в узлах не ковалентны.
Характерный пример кислых гелеобразующих полисахаридов представляют каррагинаны, содержащиеся в ряде красных водорослей. Эти полисахариды относятся к тому же типу альтернирующих структур, что и агароза, и могут быть обобщенно представлены формулой 11. Характерной и постоянной особенностью такой структуры является правильное чередование β-14иα-13-связей, но при этом структура остатков А и В варьирует в определенных пределах. Остатки А в большинстве случаев представлены 3,6-ангидро-α-D-галактопиранозой (12) или ее 2-О-сульфатом (13), но могут быть иα-D-галактопираноза-6-сульфатом (14) иα-D-галактопираноза-2,6-дисульфатом (15). Остаток В – этоβ-D-галактопираноза (16), ее 2-О-сульфат (17) и 4-О-сульфат (18) (см. с. 165).

Если отвлечься от вариаций заместителей в пиранозных циклах и рассматривать только структуры с участием 3,6-ангидро-остатков, то повторяющееся звено таких цепей (-В-А-) можно представить формулой 19. Конформационно такое звено представляет собой отрезок стержня, одна из связей которого лежит на его продолжении, а другая отходит под некоторым углом к его оси, как видно на схематической проекции 20. Такое расположение связей создает предпосылки для спирализации цепи. Действительно, в опытах со специальным образом приготовленными образцами каррагинанов, структура которых достаточно приближается к регулярной с повторяющимся звеном типа 19, при помощи рентгеноструктурного анализа было показано существование спиральной конформации цепей.
Интересно и важно для дальнейшего, что спираль оказалась двойной:две цепи с одинаковым направлением гликозидных связей тесно связаны водородными связями и закручены в главную спираль с шагом 26Å. Такая двойная спираль является довольно устойчивым образованием. Геометрические параметры каждой отдельной цепи диктуются конформацией повторяющегося звена 19 и хорошо обосновываются теоретическими методами современного конформационного анализа полимеров. Шаг этой спирали достаточно велик, так что между соседними витками остается большой промежуток («пружина» сильно растянута). В этот промежуток точно укладываются витки второй спирали;причем таким образом, что между соседними остатками из двух разных цепей возникают водородные связи, поддерживающие стабильность всей конструкции.
В свете этих данных образование трехмерной сетки из линейных молекул каррагинана, обусловливающей гелеобразование, трактуется следующим образом. В сильно разбавленном растворе (или при достаточно высокой температуре), когда межмолекулярные взаимодействия малы, форма цепей аппроксимируется конформацией беспорядочного клубка. В более концентрированных растворах (или при охлаждении) начинается образование двойных спиралей, связывающих участки разных молекул. Некоторая произвольно выбранная (и достаточно длинная) молекула может при этом образовывать несколько таких участков связывания и не с одной, а с несколькими молекулами. Тогда возникают нековалентные поперечные сшивки, и создается трехмерная сетка (рис. 10).

Теперь можно рассмотреть требования к ковалентной структуре полисахаридной цепи (в рамках общей структуры 11), соблюдение которых необходимо для возникновения такой пространственной сетки. Первым условием является возможность спирализации, для чего необходимы участки цепей из повторяющихся звеньев типа 19. Эти участки должны быть регулярными в смысле правильного чередования таких звеньев, но могут быть нерегулярными по положению и распределению сульфата. В то же время вся цепь или ее значительная часть не должнаиметь регулярную структуру из повторяющихся звеньев типа 19.
Действительно, в таком случае спирализация приводила бы к ассоциированию каждой молекулы только с одной другой молекулой (образовывалась бы непрерывная двойная спираль) и сетка не могла бы образовываться. Макроскопически это привело бы лишь к повышению кажущейся молекулярной массы полисахарида или к дальнейшей ассоциации с выпадением осадка, но не к гелеобразованию. В то же время регулярные участки не должны быть слишком короткими, так как в этом случае двуспиральные связки (узлы сетки) оказались бы чересчур слабыми или не образовывались бы совсем.
Следовательно, в структуре гелеобразующего полисахарида должны быть и регулярные последовательности типа 19, и нарушения регулярности, места, где конформация цепи резко меняется. Такую роль играют варианты звеньев типа А – сульфаты 14 и 15. Действительно, конформация дисахаридного блока –А-В- с участием (21) резко отличается от таковой для блока с 3,6-ангидрозвеном (19). Поэтому в тех точках цепи, где звено типа 12 или 13 заменено на сульфатированное звено типа 14 или 15, правильная спираль претерпевает излом (22) и связка цепей в виде двойной спирали в этом месте нарушается. Отсюда видно, что отклонения от регулярности, дефекты правильной структуры, суть не ошибки биосинтеза, как может показаться на первый взгляд, а биологически осмысленный, функционально необходимый элемент структуры.

Прочность геля и содержание в нем воды (зависящее от средних размеров ячеек) определяется в основном двумя параметрами:длиной спирализуемых и длиной неспирализуемых участков, т.е. распределением различных остатков вдоль цепи. Здесь уместно заметить, что «деспирализующие» остатки 14 и 15 являются биохимическими предшественниками 3,6-ангидрозвеньев 12 и 13. Дело в том, что биосинтез полисахаридов типа каррагинанов включает, по-видимому, сульфатирование регулярной незамещенной цепи типа…-A-B-A-B-A-B-…и замыкание ангидрозвеньев по схеме:

Таким образом, последняя стадия, осуществляемая уже на готовом полисахариде, создает гелеобразующую структуру, а степень ее протекания определяет физико-химические свойства геля. Можно полагать, что, управляя такой циклизацией, водоросли способны к тонкой адаптации своих механических характеристик к конкретным условиям среды. Например, продуцируя или активируя дополнительные количества фермента, катализирующего образование ангидроциклов, организм добивается быстрого повышения степени спирализации и, следовательно, адаптационного изменения свойств геля.
На примере каррагинанов можно проследить своеобразный характер специфичности структур таких полисахаридов. Очевидно, в этом случае не имеет большого функционального значения точная последовательность всех мономерных остатков в цепи;однако необходимым является чередование участков цепей, регулярных по альтернированию остатковβ-D-галактопиранозы и 3,6-ангидро-α-D-галактопиранозы со связями 13 и14, и участков с нарушениями такой регулярности – заменами ангидрозвеньев на соответствующие сульфаты типа 14 и 15.
Далее, функционально важно, чтобы эти участки имели определенную длину (не слишком большую и не слишком малую), но эта длина может варьировать в некоторых пределах без заметного влияния на функциональные свойства. В деспирализованных участках остатки типа 12 или 13, с одной стороны, и остатки типа 14 и 15, с другой, в известных пределах кажутся взаимозаменяемыми без заметных изменений свойств полисахарида, тогда как в спирализованных участках замена даже одного 3,6-ангидро-звена на его предшественник (сульфат 14 или 15) должна драматически сказаться на гелеобразующей способности полисахарида.
Как видно уже из структуры остатков типа А и В (12-18), число и положение сульфатных групп может варьировать в довольно широких пределах, но явно недопустимым представляется, например, наличие в остатках типа А сульфогруппы в положении 3 или ее отсутствие в положение 6 на стадии биосинтеза, предшествующей циклизации, так как из таких остатков не могли бы образовываться ангидрозвенья.
Конфигурация всех асимметрических центров в моносахаридных остатках каррагинанов строго детерминирована,
однако, как показывает строение этих полисахаридов с близкородственными по структуре и свойствам сульфатированными полисахаридами типа агара (например, с агарозой и порфираном), в остатках типа А удивительным образом оказывается возможным обращение конфигурации всех ассиметрических центров сразу:замена остатков 3,6-ангидро-D-галактозы на остатки 3,6-ангидро-L-галактозы без сущесвенного изменения функции этих полисахаридов в водорослях (такой случай «безнаказанной» взаимозаменяемости оптических антиподов, по-видимому, уникален во всей биоорганической химии). Таким образом, видно что понятие специфичности полисахаридов, по крайней мере, для разобранного примера, следует относить к типу структуры, а не к индивидуальным структурам.
В других гелеобразующих полисахаридных системах могут быть иные (и весьма разнообразные) механизмы связывания макромолекул в узлах сетки;однако характер требований к ковалентной струкуре, соблюдение которых обеспечивает выполнение обусловленных гелеобразованием функций, оказывается сходным. Так, например, в гелях альгинатов, т.е. солей альгиновой кислоты, построенной из14-связанных остатковβ-D-маннуроновой (23) иα-L-гулуроновой (24) кислот, узлы образованы кристаллитами – правильным образом упакованными участками разных молекул с регулярной структурой, подобными по упаковке кристаллическим участкам элементарных фибрилл целлюлозы. Как мы уже говорили, цепи альгиновых кислот построены по блочному принципу:в них чередуются сегменты регулярной структуры из остатков одного типа с сегментами, в которых остатки обоих типов распределены более или менее случайно. Регулярные участки, подобно целлюлозе, имеют стержнеобразную конформацию и потому способны ассоциировать в кристаллиты, а для нерегулярных участков правильная упаковка невозможна, и они образуют в сетке промежутки между узлами.
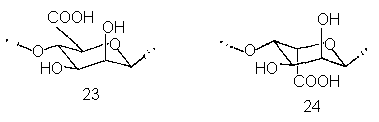
Сходный тип узлов (кристаллиты) образуется в гелях пектинов – сложных разветвленных полисахаридов, в основе молекул которых лежит цепь из β-14-связанных остатков частично этерифицированнойD-галактуроновой кислоты.
Связи цепей в кристаллитах пектинов сравнительно слабы и легко разрываются за счет гидратации моносахаридных остатков. Поэтому прочные гели образуются только при условии снижения термодинамической активности растворителя (воды) за счет растворения хорошо гидратируемых низкомолекулярных веществ (например, сахарозы). Образование пектиновых гелей в присутствии сахарозы есть физико-химическая основа ряда кондитерских проиводств, таких, как изготовление варенья, конфитюров, мармеладов и т.п. В растительных же тканях пектиновые гели служат связующим межклеточным материалом и цементирующей основой клеточной стенки.

Еще один интересный гелеобразователь практического значения – частично метилированная целлюлоза, часто называемая «метилцеллюлозой». По мономерному составу это производное характеризуется случайным распределением метильных групп:в нем есть остатки и три-О-метил-глюкозы 25, и ди-О-метил-глюкоз (например, 26), и моно-О-метил-глюкоз (например, 27), и неметилированные остатки глюкозы. Онако по условиям получения частично метилированная целлюлоза характеризуется блочным строением. Дело в том, что ее синтез выполняют путем обработки нерастворимого целлюлозного материала хлористым метилом и водной щелочью. При этом происходит сильное набухание и затем интенсивное метилирование аморфных участков микрофибрилл, тогда как фрагменты молекул, находящиеся в кристаллических участках, почти недоступны для реагентов и остаются интактными. В результате в получающемся производном чередуются сегменты цепей, характеризующихся высокими степенями метилирования, с сегментами, не метилированными совсем (или очень незначительно метилированными).
В водных растворах высоко метилированные участки разных цепей связываются гидрофобными взаимодействиями и образуют узлы сетки, а низкометилированные и потому хорошо гидратированные участки образуют межузельные промежутки. Понятно, таким образом, что гелеобразующие свойства такого производного существенным образом зависят от тонкой структуры микрофибрилл и, следовательно, от происхождения исходного целлюлозного материала. Нам представляется, что пример метилцеллюлозы, хотя и не связан непосредственно с биологической функцией полисахаридов, весьма интересен:с одной стороны, это хорошая модель биологических гелей, а с другой – образец того, как чисто технические свойства материала непосредственно зависят от надмолекулярной структуры такого чисто биологического объекта, как клеточная стенка растения – источника сырья.
Нам хочется выразить уверенность, что и другие биологические функции углеводов в обозримом будущем можно будет перевести на строгий язык ковалентных структур;причем для разного типа функций можно ожидать весьма различную по характеру структурную специфичность, совсем не обязательно сводящуюся к типовой. И тогда огромное и несколько хаотическое многообразие углеводных структур перестанет восприниматься как странная игра природы, а органично войдет как составная часть в систему представлений молекулярной биологии.
ПРИЛОЖЕНИЕ
О ЛИТЕРАТУРЕ ПО УГЛЕВОДАМ
Мировая литература по вопросам, затронутым в этой книге, насчитывает сотни обзоров и монографий, а оригинальная литература исчисляется десятками тысяч названий. Рационально выбрать из такого списка несколько десятков работ в качестве рекомендуемой литературы представлялось нам нереальным. Поэтому мы предпочли аннотировать важнейшие литературные источники, из которых читатели могут почерпнуть более полную и подробную информацию по углеводам.
Монографии
Кочетков Н.К., Бочков А.Ф., Дмитриев Б.А., Усов А.И., Чижов О.С., Шибаев В.Н. Химия углеводов. М.: Химия, 1967, 671с. Монография своего времени дает достаточно полную сводку систематизированных сведений по всей химии углеводов, а также содержит важнейшие данные по биохимии углеводов разных классов
Стоддарт Дж. Стереохимия углеводов. М.: Мир, 1975, 304 с. Рассмотрены основные аспекты стереохимии углеводов. Основные внимание уделено конформационному анализу, особенно влиянию конформационных факторов на состояние равновесий и экспериментальным методам определения преобладающих конформаций.
Бочков А.Ф., Афанасьев В.А., Заиков Г.Е. Образование и расщепление гликозидных связей. М.: Наука, 1978, 179с. Монография по химии О- и N- гликозидной связи. Подробно рассмотрены методы синтеза гликозидов, олиго- и полисахаридов. В связи с синтезом олигосахаридов проанализированы основные методы избирательной защиты функциональной групп в сахарах.
Bochkov A.F., Zaikov G.E. Chemistry if the O-glycosidic bond: Formation and cleavage. Oxford: Pergamon Press, 1979, 210p. Переработанное и значительно расширенное по сравнению с предыдущей книгой изложение химии реакций образования и расщепления О-гликозидных связей.
Обзоры
1. Advances in carbohydrate chemistry and biochemistry. New York-London: Academic Press. Ежегодник, выходящий с 1945 г. (до 1970 г. издавался под названием «Advances in carbohydrate chemistry»). Наиболее авторитетное международное издание, публикующее первоклассные исчерпывающие обзоры по
наиболее актуальным вопросам этой области знаний. Детальное изучение того или иного раздела химии и биохимии углеводов, как правило, целесообразно начинать со знакомства с соответствующими обзорами в этом издании.
The carbohydrates. Chemistry and Biochemistry. /Ed. W. Pigman, D.Horton. In 2 vols. 2-nd ed. New York – London: Academic Press, 1970-1972. Обзоры разных авторов объединены в монографию. Все обзоры написаны на самом высоком уровне и снабжены весьма представительной библиографией.
MTP International review of science. London-Baltimore: Butterworths, University of Park Press. См. разделы «Organic chemistry» и «Biochemistry». В этом многотомном издании (выходящем с 1973 года раз в два года) публикуются сжатые и чрезвычайно насыщенные информацией обзоры последних достижений в основных естественных науках. В каждой серии указанных разделов, соответствующей году, один том посвящен углеводам. Так, в седьмом томе первой серии раздела «Organic chemistry» (Ed. G.O.Aspinall. Carbohydrates, 1973) опубликованы обзоры по важнейшим вопросам химии моносахаридов и смешанных углеводсодержащих биополимеров.
Methods in carbohydrate chemistry /Ed. R.L.Whistler, M.L.Wolfrom, J.N.BeMiller. In 7 vols. New York-London: Academic Press, 1962-1976. Описаны методы выполнения, анализа, установления строения и синтеза в ряду углеводов. Каждый том содержит десятки статей разных авторов, в которых на конкретных примерах описаны те или иные методики работы с углеводами. Каждая статья предваряется очень кратким обзором по рассматриваемому вопросу. Вышедшие к настоящему времени семь томов составляют подлинную энциклопедию экспериментальной техники химии углеводов. Имеется частичный русский перевод этого издания: Методы химии углеводов. М.: Мир, 1967, 512с. (выборочный перевод, I, II, IV и V томово оригинала); Методы исследования углеводов. М.: Мир, 1975, 445с. (полный перевод VI тома).
В аналогично построенном издании «Methods in enzymology» ферментам углеводного обмена посвящены следующие тома: vol.8 (Ed. E.F.Noufeld, V.Ginsburg. New York-London: Academic Press, 1966); vol. 41,42 (Ed. W.A.Wood. New York-London, San Francisco-London: Academic Press, 1975).
Экспериментальные методы изучения крахмала освещены в книге: Рихтер М., Август З., Ширбаум Ф. Избранные методы исследования крахмала. М.:Пищевая промышленность, 1975
Обзорные работы по функционированию углеводов в живых системах регулярно публикуются в последние годы в международном журнале «Biochemica et biophysica acta», а также в журналах «Plant physiology» и «Phytochemistry». Обзоры по химии и биохимии углеводов периодически появляются в отечественных изданиях «Биоорганическая химия», «Молекулярная биология», «Успехи биологической химии», «Успехи химии».
Оригинальные статьи
Солидная доля важнейших оригинальных публикаций по химии углеводов в настоящее время приходится на международный журнал «Carbohydrate Research». Из национальных журналов,
в которых особенно часто публикуются работы по углеводам, можно отметить следующие: «Биоорганическая химия», «Журнал органической химии», «Известия АН СССР» (серия химическая), «Acta chemica scandinavica», «Canadian journal of chemistry», «Chemische Berichte», «Journal of chemical society (Perkin Translations I and II)», «Journal of organic chemistry».
Учебники
Ленинджер А. Биохимия. М.: Мир, 1976, 957с. В соответствующих главах учебника содержится сжатое, но ясное и современное изложение основ химии углеводов и гораздо более подробные сведения по их биохимии и биологической функции.
Степаненко Б.Н. Химия и биохимия углеводов. Моносахариды. М.: Высшая школа, 1977, 224 с.; Химия и биохимия углеводов. Полисахариды. М.: Высшая школа, 1978, 256 с. (Учебники для высшей школы). Большое внимание уделено биохимическим вопросам.
Популярная литература
Серия превосходных популярных статей, касающихся ряда сторон биологической функции углеводов и родственных вопросов, опубликована в сборниках: «Живая клетка» (М.: Мир, 1962, 223с.) и «Молекулы и клетки» (В 6-ти вып. М.: Мир, 1966-1977). Под ред. Г.М.Франка.
Библиография
Систематизированные библиографические указатели «Химия углеводов» (М.: Наука, 1966, 1971, 1975, 1976, 1979) охватывают основную литературу за 1961-1976 гг. Эти указатели составлены на основе картотеки по химии углеводов, систематически ведущейся в Библиотеке Института органической химии им. Н.Д.Зелинского АН СССР (Москва).
ОГЛАВЛЕНИЕ
|
Предисловие |
3 |
|
Глава 1. Структуры |
5 |
|
Что это такое? |
5 |
|
Структура моносахаридов |
7 |
|
Изображение молекул углеводов на плоскости |
12 |
|
Мутаротация |
17 |
|
Структура гликозидов |
19 |
|
Структура и разнообразие олигоахаридов |
22 |
|
Структура полисахаридов |
26 |
|
Микрогетерогенность |
38 |
|
Смешанные биополимеры |
43 |
|
Глава 2. Как устанавливают структуры |
48 |
|
Общий взгляд |
48 |
|
Мономерный анализ |
50 |
|
Установление строения моносахаридов |
55 |
|
Установление строения полисахаридов |
86 |
|
Глава 3. Синтез |
115 |
|
А зачем, собственно? |
115 |
|
Главные задачи синтеза |
118 |
|
Синтез моносахаридов |
122 |
|
Создание О-гликозидной связи |
130 |
|
Синтез олигосахаридов |
131 |
|
Глава 4. Функция |
134 |
|
Предварительные замечания |
134 |
|
Энергия. Пища. |
136 |
|
Опорные функции |
147 |
|
Маркировка поверхностей |
155 |
|
Молекулярная биология полисахаридов |
162 |
|
Приложение |
173 |
*По преимуществу (лат.)
*Характерен ишироко известен другой пример такого катализа сближением – внутримолекулярная этерификация, т.е. образование лактонов
*Хотя их традиционно называют «формами», на самом деле это истинные изомеры – соединения с разными структурами и разными свойствами, по крайней мере, если исключены или заторможены взаимные превращения. Такие изомеры носят название аномеров, сохраняемое и для других производных сахаров, где возможен тот же тип изомерии. Так, двеD-глюкофуранозы суть аномеры
*Надо сказать, что универсальным структурным элементом всех нуклеиновых кислот являетсяN-гликозидная связь остатков рибозы или 2-дезоксирибозы с основанием. Однако это связь несколько особого типа, более похожая по свойствам на О-гликозидную. Нам кажется, что связанные с такими структурами вопросы более уместно рассматривать в книге о нуклеиновых кислотах, а не в книге об углеводах
*Насколько нам известно, никто еще ни в специальной, ни в популярной литературе не анализировал сколько-нибудь подробно вопрос об информационной емкости углеводных структур
*Последовательность типа…АБАБАБАБАБ… можно записать и как (АБ)n, и как(БА)n
*Стрелкой на схеме показана гликозидная цепь, разрываемая лизоцимом после связывания с гексасахаридным отрезком полисахаридной цепи
**Строго говоря, при небольших отклонениях от нормальной структуры ферментативный гидролиз еще может происходить, но гораздо хуже
*На первый взгляд кажется, что уже в одном моле все молекулы могут оказаться различными. Однако на самом деле вероятность этого ничтожно мала. Действительно, дляNвозможных структур вероятность того, что в образце из М молекул (M<N)все молекулы будут различны, равна
![]()
Точные вычисления при N~1024трудны; однако оценка показывает, что вероятность перестает быть бесконечно малой только если М~N или М<N. Нас здесь, однако, интересуют не точные вычисления:с практической точки зрения безразлично, будет ли в образце находиться 1015или 1020молекул. В любом случае ясно, что такое вещество представляет собой смесь, абсолютно не поддающуюся разделению.
*Такую задачу способен решить рентгеноструктурный анализ, с той разницей, что конечный результат определяет положение в пространстве всех атомов сразу, и с теми ограничениями, что для этого метода требуется индивидуальное (в классическом смысле слова) вещество в кристаллическом состоянии и что даже при соблюдении этих условий при современной высоко компьютеризированной аппаратуре решение такой задачи для высокомолекулярных веществ требует весьма значительных затрат труда, приборного времени и времени ЭВМ
*В некоторых крайних случаях моносахаридный состав позволяет делать довольно уверенные заключения о нерегулярности полисахарида. Так, например, трудно предположить, чтобы полисахарид, содержащий моносахаридыA,B,C иDв соотношении100:38:4:1, имел регулярную структуру.
*Такой анализ, однако, технически сложен и поэтому мало достоверен. Дело в том, что для больших молекулярных масс это соотношение может составлять величину порядка (1:100)-(1:1000), а такие соотношения могут быть измерены экспериментально с весьма малой точностью. В разветвленных же полисахаридах молекулярная масса определяется отношением продуктов типа 2 к разности между продуктами типа 1 и типа 3. В подобных случаях результат обычно безнадежно тонет в ошибках измерений.
*Гёте И. Фауст (перевод Н.Холодковского)
*Этот интервал вообще довольно характерен для органических веществ. Дело в том, что более низкоплавкие вещества трудно бывает получить в кристаллическом состоянии, а более высокоплавкие часто плавятся с разложением, вследствие чего их «температуры плавления» перестают быть воспроизводимыми и характерными константами.
*Можно, впрочем, ограничиться и такими данными, но только если логика исследователя, условия проведения реакции, в ходе которой происходит образование изучаемого соединения, подсказывают химику структуру вещества;иными словами если кроме совпадения двух констант имеются еще какие-то независимые данные в пользу такой структуры.
*Все это справедливо, конечно, только при правильном выборе конкретного метода и условий хроматографии. Подбор подходящих условий – целая область знаний, на подробностях которой здесь останавливаться было бы неуместно. Скажем лишь, что для основных сахаров и их производных оптимальные способы хроматографического анализа уже хорошо разработаны, так что сейчас исследователю нужно не столько искать оптимумы самому, сколько внимательно просматривать оригинальную литературу
*Современная наука (в частности, химия углеводов) развивается настолько стремительно, что многие сведения, особенно о методах исследования, описываемые даже в не очень старых вузовских учебниках, уже принадлежат скорее истории, чем современности. Так, например, трудно найти учебник органической химии, в котором бы не упоминалось бы о получении озазонов для идентификации моносахаридов, хотя этот прием уже достаточно давно перестал применяться в исследовательской практике.
**Эмиль Фишер (1852-1919) обогатил науку первоклассными исследованиями в области производных пуринов, химии аминокислот и пептидов, дубильных веществ и трифенилметановых красителей. Однако особенно значительным и долговечным оказался его вклад в химию углеводов, область науки, основоположником которой его можно считать. В 1902г. Э.Фишер был удостоен Нобелевской премии «в признание выдающегося значение классических работ,связанных с сахарами и пуриновыми группами» (формулировка Нобелевского комитета)
*Иногда пик молекулярного иона в спектре отсутствует. Это осложняет расшифровку спектра, но отнюдь не делает ее невозможной
*В связи с этим, с разрешения доктора химических наук А.В.Камерницкого, воспроизведем здесь его рассказ о случае из исследовательской практики. Был снят масс-спектр некоторого вещества, предположительно – стероидного гормона. Молекулярный ион –m/e256. Вполне допустимая величина для стероидов. Следующий пик –M-32. Отщепление метанола, решили исследователи. Вполне возможно. Далее следовал пик, отвечающий иону сm/e192, т.е. М-(32x2). Отщепление второй молекулы метанола?Странно, но возможно. Затем следовалоm/e160, т.е.M-(32x3)(?!), М-(32x4),M-(32x5) и .т.д. вплоть до m/e32! Иными словами, спектр отвечал последовательному отщеплению семи молекул метанола и образованию в остатке восьмой молекулы метанола, причем никаких других пиков в спектре не было. Стероид, состоящий из одного метанола?
На самом деле вещество оказалось элементарной серой (S8), а его спектр описывал распад с последовательным отщеплением атомов серы(AB 32). Так что не всегда трактовка структуры первичных ионов и отщепления простых молекул бывает однозначна
*И не только полисахаридов, а вообще, для решения структурно-аналитических задач в тех весьма сложных многочисленных случаях, когда вещества находятся в сложных смесях, а количество материала жестко ограничено. Характерно в этом отношении, например, что хромато-масс-спектрометр был установлен на американской автоматической межпланетной станции «Викинг», главной задачей которой является поиск органических веществ и жизни на Марсе.
*Можно, например, указать на две книги:Бранд Дж., Элингтон Г. Применение спектроскопии в органической химии (М.:Мир, 1967) и Маклочан К.А. Магнитный резонанс (М.: Химия, 1976)
*В спектрах, публикуемых в научных статьях, над пиками действительно обычно пишут, к каким протонам они относятся. Однако это уже результат трактовки исходного, получаемого с прибора «немого» спектра
*Исключение могут составлять синглеты, т.е. сигналы протонов, находящиеся вне системы спиновых связей и потому независимых от остальных. Примером, характрным для химии углеводов могут быть сигналы ацетильных групп – синглеты с химическим сдвигом около 2 м.д.
*Разумеется, человек, сидящий за прибором, сам может быть первоклассным исследователем, а не только оператором. Но чаще его роль все же техническая:получить от химика вещество и квалифицированно снять его спектр. Вообще же взаимоотношения органик-физико-химик составляют сложную научно-этическую проблему, заслуживающую специального обсуждения
*На самом деле для этого применяют несколько более сложную, но аналогичную деструктивную реакцию. Наше упрощенное изложение не искажает, однако, принципиальную сторону дела
*Заметим, что такой опосредованный вывод о последовательности не позволяет судить о конфигурации гликозидной связи 3,6-ангидро-L-галактопиранозы (в отличие от остаткаβ-D-галактопиранозы, конфигурация которого непосредственно следует из структуры агаробиозы).
*В действительности цепи агарозы высоко регулярны – дальний порядок не нарушен. Однако эти сведения получены всей совокупностью методов структурного анализа, примененных к агарозе, и не могли быть извлечены только из результатов частичного гидролиза описанного типа
*Точнее говоря, аддитивным является не удельное вращение, а молекулярное, т.е. вращение, отнесенное не к весовой, а к молярной концентрации вещества
*Шашков А.С., Усов А.И., Яроцкий С.В. – Биоорганическая химия, 1978, т.4, с.74
*Выделение и очистка ферментов, даже из доступного источника – работа весьма трудоемкая и требующая высокой квалификации, а изучение их специфичности (знание которой, собственно, и обеспечивает столь высокую информативность ферментативного гидролиза) составляет сложную самостоятельную исследовательскую работу. Что же касается высоко очищенных ферментных препаратов, выпускаемых промышленностью, то многие из них исключительно дороги.
*Литературная газета, 1973, 28 февраля
*Вудворд Р.Б. Синтез. – В кн.:Перспективы развития органической химии. М.: ИЛ, 1956, с. 121
*Вудворд Р.Б. Синтез.- В кн.:Перспективы развития органической химии. М.:ИЛ, 1956, с.119
*Говоря в этой части об углеводах, мы имеем в виду прежде всего моносахариды, полисахариды и более сложные биополимеры, содержащие полисахаридные и олигосахаридные цепи, и традиционно не включаем в это понятие нуклеиновые кислоты, хотя в состав любого нуклеотида обязательно входит остатокD-рибозы или 2-дезоксиD-рибозы
*Ленинджер А. Биохимия. М.: Мир, 1976, с. 11-12.
*Мелвилл Г. Моби Дик, или Белый кит. М.: Географгиз, 1962, с.311
**Махабхарата. Книга пятая. Ульйогапарва. Л.:Наука, 1976, с.160
***Нараян Р.К. Боги, демоны и другие. М.: Наука, 1975, с.244
*Это условие можно вывести из самых общих законов химии
*Такого рода патологические отклонения в структуре гликогена изредка случаются:это так называемые гликогеновые болезни, приводящие к тяжелейшим нарушениям углеводного обмена и обычно кончающиеся гибелью больного в раннем возрасте.
*Слово «специфичность» применительно к биомолекулам, вообще говоря, не строгий термин, а скорее эпитет, который часто употребляется в биологической литературе, но которому разные авторы придают несколько разный смысл.
*Названия «первичная» и «вторичная» связаны с последовательностью биосинтеза этих частей клеточной стенки
*По другим данным сечение элементарной фибриллы – 30Х25Ǻ, а число цепей в нем 32
*Видершайн Г.Я. – Молекулярная биология, 1976, т.10, с.957