

Глава 3
Синтез
А зачем, собственно?
В естественных науках вопрос «зачем?» принадлежит к числу наиболее каверзных. С одной стороны, в ответе на него (если, конечно, ответ дан вдумчиво и серьезно) заключена конкретная стратегия исследования, программа, направляющая и концентрирующая усилия ученых. С другой стороны, в слишком прямолинейном ответе, во всяком случае в его практическом воплощении, заключена определенная опасность: он отсекает зарождающиеся новые направления науки, перспективы которых сегодня еще не ясны, но которые завтра могут оказаться весьма плодотворными. Гениальный Вудворд высказался на этот счет весьма категорически:«Я не стану скрывать мою уверенность, хотя она, может быть, и идет вразрез с зачастую слишком утилитарным духом времени: синтез ради синтеза будет продолжаться. Органический синтез увлекателен, полон приключений и опасностей, он часто требует высокого искусства. Одного этого достаточно – органическая химия была бы значительно скучнее, если бы ни одна из ее частей не содержала такого стимула»*.
Мы никоим образом не оспариваем этого мнения, тем более, что из вудвордовских «синтезов ради синтеза» развились целые направления органической химии весьма общего значения. Однако применительно к углеводам синтез как самодовлеющее искусство не получил значительного распространения. Напротив, большинство синтетических исследований по углеводам подчинено (явно или неявно) решению одной общей проблемы, выходящей за рамко собственно синтеза. Более того, синтез играет в ней скорее подсобную роль. Поэтому для того, чтобы представить себе суть стратегии синтетических исследований
в химии углеводов, нужно прежде всего осознать их конечные цели.
Мы уже говорили о том, что химические исследования в химии углевродов направлены в конечном счете преимущественно на решение проблемы «структура-свойство», «структура-функция». Один подход к решению – это выделение индивидуальных природных углеводов, установление их структуры и сопоставление полученных сведений с наблюдаемыми свойствами природных углеводов и их биологическими функциями. На этой основе можно сделать те или иные выводы и обобщения о роли структурных факторов в проявлении тех или иных свойств природных соединений. Однако для того, чтобы подтвердить такие звключения, а также для того, чтобы их уточнить и детализировать, очень важно иметь возможность варьировать структуру изучаемых соединений, варьировать целенаправленно, плавно изменяя в них какой-то определенный структурный параметр. И здесь незаменимым оказывается точный синтез как источник модельных объектов исследования с заданной структурой.
Сам по себе природный объект, например полисахарид или смешанный углеводсодержащий биополимер, часто бывает столь сложным, что непосредственно понять его свойства и функцию на молекулярном уровне современной науке оказывается не под силу. И тут неоценимую помощь оказывают упрощенные модели такого полимера, включающие определенные элементы его структуры. Такую роль, например, играют олигосахариды по отношению к полисахариду или полисахаридные цепи гликопротеина по отношению к природному гликопротеину. Источником подобных упрощенных систем может служить, с одной стороны, сам исходный биополимер, а с другой – их химический синтез.
Другим случаем моделирования является синтез субстратов ферментов углеводного обмена и их структурных модификаций, служащих для изучения субстратной специфичности таких ферментов или для их избирательного ингибирования.
Универсальным приемом исследования в молекулярной биологии и примыкающих к ней биологических дисциплинах сейчас является использование природных соединений, меченных радиоактивными изотопами. С его помощью удается решить огромное число типов задач, связанных с прослеживанием путей превращений тех или иных веществ
в живых системах и в их упрощенных экспериментальных моделях. Химический синтез – мощное средство получения природных соединений и их структурных аналогов с определенным, заранее заданным положением меченых атомов в молекуле.
Еще одна традиционная задача органического синтеза – подтверждение строения природных соединений путем так называемого встречного синтеза. Смысл его в том, что наиболее надежным, бесспорным доказательством установленной аналитическими методами структуры нового вещества является его химический синтез и идентификация природного и синтетического образцов. Значение такого подхода сейчас, правда, несколько снижается благодаря развитию высокоточных и чрезвычайно надежных методов структурного исследования, что иногда делает встречный синтез не столь обязательным, иногда даже украшательским элементом исследования. Тем не менее во многих случаях необходимость его не подлежит сомнению.
Наконец, еще один, не столь может быть очевидный источник интереса к синтетическим исследованиям в биоорганической химии, и, в частности, в химии углеводов, состоит в следующем. Дело в том, что органический синтез был и остается до сих пор наиболее эффективным и всеобъемлющим способом изучения химии самих объектов синтеза и наиболее строгим способом проверки наших представлений о связи структуры соединений с их химическими свойствами и поведением. Здесь опять уместно процитировать Р.Б.Вудворда, который говорил:«Вряд ли можно отрицать, что успешный исход синтеза, состоящего более чем из 30 стадий, является суровым испытанием способности науки к предвидению, а также проверкой ее познавательной мощи в сфере изучаемых объектов»*. Хотя эти слова написаны более четверти века назад, они сохраняют свое значение и поныне и в полной мере применимы к химии углеводов.
Общий вывод, который следует из приведенных рассуждений, сводится к тому, что органический синтез в области углеводов должен быть направлен (и направлен в действительности) в первую очередь на создание природных и аналогичных им структур. А это создает основу для
формулирования специфической проблематики этой области и классификации основных принципов и методов синтетической химии углеводов
Главные задачи синтеза
Итак, синтез природных углеводных структур и их ближайших аналогов. В первую очередь, это синтез моносахаридов, природных гликозидов, олигосахаридов и полисахаридов. Олиго- и полисахариды, как мы помним, построены из остатков моносахаридов, соединенных О-гликозидными связями. В природных гликозидах с тем же типом связи моносахаридные остатки соединены с неуглеводными агликонами. Поэтому в синтезе олиго- и полисахаридов или гликозидов задача химика сводится в конечном итоге к тому, чтобы соединить моносахаридные остатки друг и другом или с агликоном гликозидными связями.
Таким образом, в синтезе природных углеводов и их аналогов можно выделить две общие и наиболее фундаметальные проблемы:синтез моносахаридов и построение гликозидных связей.
В понимании химика XIXв. «синтезировать органическое соединение» означало получить его искусственно из наиболее простого сырья, в идеале – из элементов. Даже если исходными служили другие органические соединения (например, уксусная кислота или этилен), конечный результат также мог рассматриваться как полный синтез, т.е. синтез из элементов, поскольку способы получения этих исходных соединений из элементов уже существовали. В современном тонком органическом синтезе исследователь, как правило, не задумывается о том, насколько далеко отстоят используемые им исходные соединения. Важно только, чтобы эти исходные соединения были достаточно доступны:производились промышленностью, либо, по крайней мере, чтобы способы их получения (необязательно путем синтеза) были описаны в литературе ранее. Отсюда понятно, что при синтезе олигосахаридов, полисахаридов и гликозидов основными исходными соединениями служат составляющие их моносахариды и их подходящие производные. Что же касается синтеза самих моносахаридов, то его общие принципы заслуживают специального обсуждения.
Построение характерного для большинства моносахаридов прямого углеродного скелета из пяти-шести углеродных атомов не составляет проблемы для современной органической химии. Несколько сложнее, но также вполне в пределах синтетических возможностей, снабдить каждый из этих атомов функциональной группой – спиртовой, аминогруппой, карбонильной и т.д. Еще Бутлеров более 100 лет назад осуществил синтез смеси моносахаридов с присущей им бутлеровской структурой, использовав одно из простейших органических соединений – формальдегид. Загвоздка, однако, заключается в том, что большинство углеродных атомов моносахаридной молекулы асимметрично. Поэтому синтез природного моносахарида предполагает не только создание нужного углеродного скелета, но и возможность придания всем асимметрическим центрам вполне определенной относительной и абсолютной конфигурации. А такая задача весьма трудна даже для современной высокоразвитой органической химии, если в качестве исходных соединений используются простые молекулы без элементов асимметрии или даже более сложные системы, содержащие один-два асимметричных центра с нужной конфигурацией.
Поэтому гораздо более простым и эффективным оказывается такой путь:проведение совокупности последовательных трансформаций доступных моносахаридов, целенаправленно приводящее к другому моносахариду с желаемой структурой. Доступными (выпускаемыми промышленностью) моносахаридами, чаще всего используемыми в качестве исходных соединений в таких синтезах, являютсяD-глюкоза,D-галактоза,D-манноза, D-иL-арабиноза,D-ксилоза,N-ацетил-D-глюкозамин,D-фруктоза,D-рибоза и немногие другие. Следующие несколько примеров могут проиллюстрировать такие «трансформационные» принципы синтеза.
Для получения D-глюкуроновой кислоты (1) изD-глюкозы (2) необходимо окислитьCH2OH-группу до карбоксила;чтобы получить 4-О-метил-D-глюкуроновую кислоту (3), нужно, кроме того, прометилировать гидроксил при С-4;для синтеза 3-амино-3-дезокси-D-глюкозы (4) – заменить гидроксильную группу при С-3 на аминогруппу. Для синтеза 2-дезокси-D-рибозы (5) изD-рибозы (6) илиD-арабинозы (7), или для синтезаD-фукозы (8) из D-галактозы (9)
необходимо в исходном моносахариде удалить соответствующую гидроксильную группу. Синтез D-ликсозы (10) изD-ксилозы (11) требует обращения конфигурации одного асимметричного центра приC-2, а изD-арабинозы (7) – при С-3 и т.п.

Не следует думать, что наиболее удобный путь синтеза – это всегда минимальная трансформация исходного соединения. В этом смысле приведенные схемы нужно рассатривать только как наглядную иллюстрацию принципи. Тем не менее они содержат примеры основных типов трансформаций, используемых в реальных синтезах моносахаридов, таких, как окисление или восстановление одного из углеродных атомов исходного моносахарида, замена гидроксила на иную функцию (аминогруппу, метокси-группу и т.п.), обращение конфигурации. Сюда можно
добавить еще несколько типичных превращений: удлинение углеродной цепи, введение разветвлений, а также одновременное или последовательное выполнение нескольких подобных трансформаций. Химические реакции, приводящие к характерным для синтеза моносахаридов изменениям в структуре молекулы, относительно просты и применительно к простым органическим соединениям, выполняются обычно без особых затруднений.
Синтез моносахаридов из моносахаридных же предшественников удобен, разумеется, тем, что большая часть целевой структуры уже имеется в исходном соединении:весь углеродный скелет или, по крайней мере, его значительная часть, большинство функциональных групп, нужная конфигурация большинства асимметрических центров. Однако именно в этом и заключается главная трудность. Ведь для того, чтобы выполнить целенаправленную трансформацию, нужно суметь не затронуть другие, химически весьма близкие группировки в исходной молекуле. Например, при синтезе 4-О-метил-D-глюкуроновой кислоты (3) нужно тем или иным способом обеспечить метилирование именно четвертого гидроксила, а не весьма сходных с ним по химическим свойствам третьего или второго. Аналогично, для превращенияD-ксилозы (11) вD-ликсозу (10) надо добиться обращения конфигурации углеродного атома С-2, несущего в пиранозной форме экваториальный вторичный гидроксил, и не затронуть при этом конфигурацию двух других центров (С-3 и С-4), также находящихся в пиранозном цикле и также связанных с экваториальными вторичными гидроксилами. Аналогичные задачи возникают в каждом из приведенных выше принципиальных синтетических путей и практически в каждом другом синтезе моносахаридов по схеме трансформаций.
Проблема обеспечения избирательности реакции при наличии в молекуле нескольких реакционных центров – проблема региоспецифичности (от латинского regio – область) – имеет в органической химии весьма общее значение, но особенно характерна именно для химии углеводов, небольшой углеродный скелет которых до предела насыщен однотипными функциональными группами, создающими вместе с гликозидным центром неповторимый и весьма колоритный ансамбль. Как же может быть решена задача?
Ключевым принципом, позволяющим добиваться высокой, даже абсолютной региоспецифичности таких реакций, является применение защитных групп. Этот подход настолько важен, что заслуживает специального рассмотрения.
Синтез моносахаридов
Защитные группы
Сама по себе идея применения защитных групп широко известна в общей органической химии. Вот классический пример. Нужно пронитровать аналин и получить п-нитроанилин. Азотная кислота – сильный окислитель, а аналин легко окисляется. Следовательно, нитровать его непосредственно нельзя. Поэтому аминогруппу анилина предварительно защищают:превращают в ацетат, гораздо более устойчивый к окислителям, затем нитруют и в заключение удаляют защиту с аминогруппы щелочным гидролизом:

Здесь все просто. Анилин содержит два весьма различных по характеру реакционных центра – аминогруппу и ароматическое ядро. Поэтому избирательно защитить один из них не составляет проблемы. Продукт реакции – п-нитроанилин – весьма устойчивое соединение и легко переживает условия достаточно жесткого щелочного гидролиза. Следовательно, удаление защиты также не вызывает затруднений. В химии углеводов дело обстоит несравненно сложнее. Прежде всего, здесь функциональные группы весьма сходны, так что ввести защиту избирательно – а в этом весь смысл такой операции – весьма непросто. Таких групп в молекуле несколько (чтобы не сказать много), а защитить нужно все, кроме одной-двух. Понятно, что это обстоятельство, вообще говоря, не упрощает задачу. Наконец, сами углеводы и практически все их производные – соединения достаточно высоко реакционноспособные. Из-за этого возможности воздействий,
пригодных для удаления защит на заключительных стадиях, а следовательно, типы применяемых защитных групп жестко ограничены.
Основные требования к защитным группым достаточно очевидны. Во-первых, они должны допускать избирательное введение. Во-вторых, сами защиты должны быть вполне устойчивы в условиях основной реакции. В-третьих, защиты должны допускать удаление в условиях, обеспечивающих сохранность как самой углеводной структуры, так и, разумеется, результатов главной реакции, ради осуществления которой и возводились защитные сооружения. Наконец, не столь принципиально, но весьма немаловажно, чтобы реакции введения и удаления защитных групп проходили с высокими выходами:иначе весь многостадийный синтез будет сопряжен со слишком значительными потерями.
Из всего перечисленного наибольшие затруднения вызывает избирательное введение. Здесь нет каких-то разработанных правил, следуя которым можно механически выбрать необходимую последовательность превращений и типы защитных групп. Тем не менее есть ряд хорошо разработанных реакций, ведущих к образованию защит, и ряд принципов обеспечения их региоспецифичности. Так что сейчас грамотый синтетик может составить реальный план синтеза, ведущего к избирательному освобождению любой функциональной группы в любом моносахариде. Но, подчеркнем еще раз, это не механическое применение готовых правил, а творческий процесс, требующий тщательного учета задач конкретного синтеза и выбора оптимальной схемы из ряда возможных. Поэтому не будем пытаться дать, так сказать, алгоритм для избирательной защиты функций, а опишем лишь некоторые элементарные приемы, применяемые в химии углеводов для этой цели.
Рассмотрим D-глюкозу (2). Пусть нам надо защитить все гидроксильные группы, кроме гидроксила при С-6. Такая задача сравнительно проста, так как интересующий нас гидроксил первичный и заметно отличается по реакционной способности от остальных гидроксилов в молекуле – вторичных спиртовых и полуацетального. Эту повышенную реакционную способность и используют на ключевой стадии синтеза. Глюкозу обрабатывают трифенилметилхлоридов (тритилхлоридом, как его часто сокращенно называют) в пиридине. При реакции тритилхлорида
со спиртами образуются простые тритиловые эфиры. Тритильная группа весьма объемиста, поэтому тритилирование пространственно более затрудненных вторичных спиртов проходит медленно, тогда как первичные тритилируются легко. Благодаря этому тритилирование глюкозы проходит с высокой избирательностью и ведет к образованию тритилового эфира 12. Все остальные гидроксилы можно далее защитить ацетилированием уксусным ангидридом в пиридине. В полученном производном 13 все функциональные группы защищены, но защищены по-разному. Тритиловый эфир может быть разрушен кислотным гидролизом в таких условиях, которые не затрагивают сложные эфиры – ацетаты. Продуктом такого гидролиза является тетраацетат 14, в котором свободен единственный гидроксил – при С-6.

Обратите внимание, каким парадоксальным путем идет этот синтез: для того, чтобы избирательно освободить гидроксил при С-6, мы начинаем с того, что его защищаем. И тем не менее конечная цель достигается весьма успешно. Пример характерен в двух отношениях: во-первых, химия углеводов в части логики введения избирательных защит полна таких парадоксов, а во-вторых, использование избирательного тритилирования является общим (что редко в этой области) методом освобождения первичного гидроксила в сахарах.
Другой участок в молекуле моносахарида, также обладающий специфическими свойствами,- это гликозидный центр. Для его избирательной защиты чаще всего применяют синтез низших гликозидов, в простейшем случае
путем катализируемой кислотами конденсации моносахаридов со спиртами (синтез гликозидов по Фишеру). Наиболее распространенные производные для этой цели – метилгликозиды, каковы, например α-метил-D-глюкопиранозид (15),α-метил-L-рамнопиранозид (16) илиβ-метил-L-арабинопиранозид (17). Для расщепления метил-гликозидов необходимо произвести достаточно жесткий кислотный гидролиз или ацетолиз, что не всегда приемлемо по условиям устойчивости основного продукта. Чтобы избежать этого осложнения, пользуются бензил-гликозидами[например,β-бензил-D-галактопиранозидом (18)], в которых защита может быть удалена в специфических условиях путем гидрогенолиза над палладиевым катализатором (см. схему).

Наибольшие трудности возникают при необходимости избирательной защиты части вторичных гидроксилов моносахаридов, так как эти группы обладают наиболее близкими химическими свойствами. Чаще всего ключевой стадией в таких синтезах является образование тех или иных ацеталей или кеталей. Как известно, альдегиды и кетоны способны легко конденсироваться со спиртами в присутствии кислотных катализаторов с образованием ацеталей или кеталей 19. Если в реакцию вводят двухатомный спирт с подходящим расположением гидроксильных групп, то такая реакция приводит к аналогично построенным циклическим производным типа 20. Ацетали и кетали расщеаляются кислотным гидролизом в сравнительно мягких условиях и весьма устойчивы к щелочам, что делает их пригодными
в качестве защитных групп в многочисленных типах синтезов.

Для того, чтобы циклические производные типа 20 могли образовываться достаточно легко, необходимо соблюдение определенных требований к структуре исходного двухатомного спирта. Две его гидроксильные группы не должны быть расположены слишком далеко одна от другой, так как в противном случае вероятность замыкания цикла резко падает и реакция идет предпочтительно межмолекулярно с образованием линейных олигомеров. Кроме того, возникновение циклической системы не должно вызывать значительных дополнительных напряжений в остальной части молекулы.
По этим причинам возможность образования циклических ацеталей или кеталей подчиняется жесткому контролю со стороны всей структуры, стереохимии и конформации субстрата. В результате реакции, ведущие к таким алкилиденовым производным, протекают весьма избирательно и затрагивают не все, а лишь вполне определенные гидроксильные группы моносахарида или его частично защищенного производного. Таким образом, введение алкилиденовых группировок позволяет резко нарушить монотонность функциональных групп исходных соединений и создает основу для разнообразных способов избирательной защиты спиртовых гидроксилов.
Трансформирующие реакции
Проблема региоспецифичности реакций моносахаридов, как мы видели, может быть разрешена при помощи системы защитных групп. Помимо этого при направленных трансформациях моносахаридов необходимо еще обеспечить их стереоспецифичность, так как в большинстве случаев такие реакции протекают у одного из асимметрических центров и приводят к соединениям, в которых новая группировка также связана с асимметрическим атомом углерода. В целом среди важнейших типов органических
реакций наибольшими возможностями, с точки зрения обеспечения стереоспецифичности, обладают ионные реакции, т.е. те, которые протекают через заряженные или высоко поляризованные промежуточные продукты или переходные состояния. Именно такие реакции являются главными инструментами при работе химика-синтетика с углеводами.
Одним из наиболее употребительных типов реакций в этой области являются реакции нуклеофильного замещения при одном из углеродных атомов производного моносахарида. Вот общая схема протекания таких реакций:
![]()
Результатом реакции является обмен заместителей при центральном углеродном атоме. Реагентами служат отрицательно заряженные или нейтральные частицы, имеющие неподеленную пару электронов, так называемые нуклеофилы, такие, например, как HO:-, :CN-, CH3COO:-, N3-, R3N:и т.п. При реакции связь С-Х разрывается таким образом, что уходящая группа Х уносит электронную пару, составляющую ковалентную связь. Для того, чтобы реакция протекала достаточно эффективно, важно, в частности, чтобы Х была, как говорят, хорошо уходящей группой, т.е. имела бы достаточно большое сродство к электрону и образовывала бы достаточно стабильную частицу с неподеленной электронной парой. Хорошие уходящие группы – это те, например, которые при отщеплениии дают высоко стабильные анионы (Cl-, Br-, CH3SO2O-и т.п.)
Спиртовые гидроксилы – основной тип функции в моносахаридах – по ряду причин являются плохими уходящими группами в нуклеофильном замещении. Поэтому их обычно модифицируют, превращая в эфиры сульфокислот (сульфонаты), чаще всего в эфиры метаносульфокислоты (мезилаты, на схемах обычно обозначаемые знаком Ms) или п-толуолсульфокислоты (тозилаты, обозначаемые обычноTs). Остаток сульфокислоты – хорошая уходящая группа, легко замещаемая во множестве реакций нуклеофильного замещения, а сам синтез мезилатов и тозилатов выполняется весьма просто, обычно обработкой спирта соответствующим хлорангидридом в пиридине по схеме:
![]()
Большое разнообразие нуклеофилов, способных замещать сульфонилоксигруппы в сахарах по механизмуSN2, открывает богатейшие синтетические возможности. Так, например, замещение ацетатом или бензоатом приводит к соответствующим легко омыляемым эфирам, т.е. ведет в конечном счете к обращению конфигурации одного асимметрического центра;замещение на азид с последующим восстановлением позволяет синтезировать аминодезоксисахара; замена на иод или серу ведет к галогендезокси- и к тиосахарам.
Как известно, спирты сравнительно легко окисляются: первичные до карбоновых кислот, а вторичные – до кетонов.
![]()
Окисление спиртовых групп в производных моносахаридов по таким схемам широко применяется в синтетической практике. Окисление первично-спиртовой группы служит для синтеза уроновых кислот.
Введение кетогруппы в производные моносахаридов открывает богатейшие синтетические возможности, связанные с чрезвычайно многообразной реакционной способностью карбонильной группы. Основным путем получения таких кетонов служит оксиление производных, содержащих одну вторичную гидроксильную группу. В сравнении с обычными спиртами вторично-спиртовые группы в сахарих поддаются окислению с некоторым трудом. Поэтому для этих целей приходится применять энергичные окислители, такие, как четырехокись рутения или комбинация диметилсульфоксида с реагентами типа ангидридов (уксусный ангидрид, P2O5, дициклогексилкарбодиимид и некоторые другие). Несмотря на некоторую экзотичность этих окислителей, их широко применяют в химии углеводов. Такие методы дают сейчас синтетику возможность окисления практически любой вторично-спиртовой группы и, следовательно, введения карбонильной функции в почти любое желаемое положение.
Восстановительные реакции в химии углеводов употребляются главным образом для введения дезоксизвена и для перехода от карбонильных производных к спиртам, а также для удаления бензильной защиты. Каталитический гидрогенолиз галоген-дезоксисахаров, главным образом иодидов, а также тиопроизводных, ведет к образованию
дезоксисахаров по схеме:
![]()
Для синтеза 6-дезоксигексоз обычно применяют ионное восстановление тозилатов алюмогидридом лития (вторичные тозилаты восстанавливаются иначе).
Восстанавливая карбонильную группу комплексными гидридами металлов, например боргидридом натрия, можно получить соответствующие спирты. В циклических кетонах направление подхода реагента и, следовательно, конфигурация образующегося спирта контролируются пространственными факторами, что нередко может обеспечить высокую стереоспецифичность реакции.
Если нужно удлинить цепь углеродных атомов исходного моносахарида или внести в нее разветвления, то возникает необходимость в образовании новых С-С-связей. Наибольшее значение для достижения этих целей имеют реакции карбонильных групп моносахаридов и их производных с реагентами, содержащими нуклеофильный атом углерода, такими, как магнийорганические соединения (например, 87), синильная кислота (88), диазометан (89), илиды (например, 90) и т.п. Общая особенность этих соединений состоит в том, что их можно рассматривать, по крайней мере формально, как доноры карбанионов: частиц с неподеленной электронной парой и отрицательным зарядом на атоме углерода.
Карбонильная группа сильно поляризована: электроны двойной связи оттянуты к атому кислорода, вследствие чего на нем сосредоточен частичный отрицательный заряд, а на атоме углерода – частичный положительный. Поэтому карбонильная группа легко подвергается нуклеофильной атаке, неизменно направленной на ее углеродный атом. В случае соединений с нуклеофильным атомом углерода, как иногда говорят, С-нуклеофилов, такие реагенты приводят к образованию новых С-С-связей.
Конденсация кетопроизводных моносахаридов, содержащих карбонильную группу в циклической системе, может протекать с высокой стереоселективностью, если другие заместители создают значительную неэквивалентность пространственного окружения «верхней» и «нижней» стороны карбонильной группы. В подобных случаях подход реагента осуществляется предпочтительно со стороны
наименее экранированной другими группами в молекуле, что и обусловливает образование нового асимметрического центра с определенной конфигурацией. На таких реакциях основаны многие синтезы моносахаридов с разветвленной цепью.
Для синтеза высших сахаров с неразветвленной углеродной цепью применяют сходные реакции С-нуклеофилов с альдегидными группами альдоз. Один из наиболее старых методов в этой области – циангидринный синтез, о котором мы уже говорили в связи с работой Фишера по установлению конфигурации моносахаридов. Использование в подобных реакциях производных моносахаридов с защищенными спиртовыми гидроксилами и свободной альдегидной группой, так называемых аль-форм сахаров значительно расширяет круг применимых реагентов и, следовательно, синтетические возможности реакций. Аналогичное применение находят производные моносахаридов с защищенным гликозидным центром и содержащие альдегидную группу на противоположном конце углеродной цепи.
Создание О-гликозидной связи
Пожалуй, наиболее важной чертой гликозидной связи с точки зрения синтетика или, по крайней мере, чертой, доставляющей ему наибольшее количество хлопот, является изомерия гликозильного остатка. Как мы помним, типичный моносахарид может образовывать четыре изомерных гликозильных остатка и, следовательно, четыре типа гликозидной связи: α- иβ-аномеры пиранозной формы иα- иβ-аномеры фуранозной формы. Потому при синтезе определенного гликозида из данного моносахарида и данного спирта необходимо добиться не только создания нужной – гликозидной – связи (это общая задача любого синтеза), но и обеспечить образование гликозильного остатка с определенным размером цикла, а также обеспечить определенную конфигурацию гликозидного центра, т.е. добиться стереоспецифичности или хотя бы стереоселективности реакции.
Все современные подходы к гликозидному синтезу строятся на одном общем принципе – на нуклеофильном замещении при гликозидном центре, причем нуклеофилом выступает агликон – либо как таковой, в виде гидроксилсодержащего соединения, либо в виде некоторых
производных, содержащих при атоме кислорода катионоидную уходящую группу. Для эффективного протекания такой реакции необходимо, чтобы в молекуле исходного производного моносахарида – гликозилирующего агента – содержалась хорошая уходящая группа при гликозидном центре, т.е. чтобы последний был активирован к нуклеофильному замещению. Таким образом, в самом общем виде эти реакции можно описать следующей схемой:
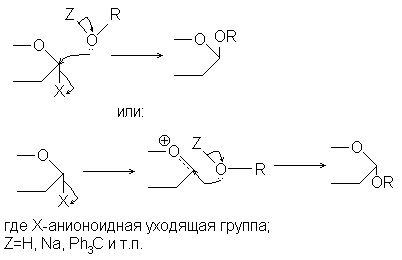
Во всех современных методах гликозидного синтеза применяют гликозилирующие агенты, в которых все спиртовые гидроксилы защищены. Этим достигается сразу два результата. Во-первых, исключается самоконденсация – гликозилирование собственных гидроксильных групп. Во-вторых, защита спиртовых гидроксилов закрепляет циклическую систему производного моносахарида, исключает изомеризацию гликозильного остатка (типа мутаротации) и обеспечивает образование гликозида с определенным, заранее заданным размером цикла. Чаще всего для этой цели используют сложноэфирную защиту, например, ацетаты, легко удаляемые мягким щелочным сольволизом (гидролизом или метанолизом), который не затрагивает обычные гликозидные связи. Для этой же цели применяют бензильную защиту – простые бензиловые эфиры расщепляются каталитическим гидрогенолизом, к которому гликозидные связи инертны.
Синтез олигосахаридов
Мы уже неоднократно упоминали о том, что олигосахариды могут служить удобными, в некоторых случаях идеальными моделями полисахаридов, с помощью которых удается относительно легко выяснить многие вопросы химии и биохимии их более сложных прототипов – самих полисахаридов. Действительно, типичный олигосахарид – это в полном смысле слова «маленький полисахарид»;и все те особенности структуры и свойств полисахаридов, которые не связаны специфическим образом с их высокомолекулярностью, в полной мере обнаруживаются и для олигосахаридов. Более того, в большом классе биологических явлений, включающих взаимодействие полимеров один с другим и даже клеток друг с другом, нередко определяющим фактором оказываются контакты поверхностных участков, представляющих собой невосстанавливающие концы полисахаридных цепей.
Часто в таком взаимодействии существенным оказывается только соприкосновение с коротким – олигосахаридным – концевым участком, а не со всей макромолекулой в целом. Поэтому олигосахариды, структура которых повторяет структуру такого концевого участка большой молекулы, с большой полнотой моделирует многие биологические свойства самого биополимера. Отсюда становится понятным, что синтезу олигосахаридов исследователи уделяют большое внимание, причем интерес к этой проблеме особенно обострился в самые последние годы в связи со значительным прогрессом в познании биологических функций полисахаридных структур. В результате во всей проблеме гликозидного синтеза синтез олигосахаридов сейчас прочно занимает центральное положение.
По сравнению с синтезом сложных гликозидов других классов синтез олигосахаридов ставит перед исследователем ряд дополнительных задач, связанных с обеспечением региоспецифичности реакций в агликоновой части будущей молекулы. В этом отношении наиболее простой случай представляет собой синтез дисахаридов. Для его выполнения надо решить две задачи: обеспечить введение в молекулу гликозильного остатка с нужным размером цикла и нужной конфигурацикй гликозидной связи и обеспечить гликозилирование определенного гидроксила в моносахаридном остатке, играющем роль агликона.
Для того, чтобы добиться региоспецифичности гликозилирования агликоновой компоненты, прибегают к частично защищенным производным сахаров, содержащим одну гидроксильную группу. В синтезе высших олигосахаридов возникает еще одна задача: создание нужной последовательности моносахаридных остатков. Наконец, нужно упомянуть о проблеме синтеза полисахаридов. При всех достоинствах олигосахаридов как моделей природных полисахаридов работа с синтетическими полисахаридами принципиально допускает, разумеется, большее приближение к природным прототипам и, что еще важнее, позволяет осуществить гораздо более тонкое направленное варьирование их структуры. Однако трудности в разработке методов синтеза полисахаридов весьма значительны, а успехи пока еще очень скромны. Тем не менее есть основания надеяться, что в этой области науки заканчивается индукционный период развития и в обозримом будущем в ней можно ожидать существенного прогресса
* * *
Мы рассмотрели – крайне схематически – основные задачи синтеза в ряду углеводов и лишь упомянули принципы, лежащие в основе современных решений этих задач. Сколько-нибудь подробное изложение вопроса не может быть выполнено в рамках настоящего издания – ему должна быть посвящена отдельная книга. Здесь мы можем лишь подчеркнуть, что синтез в углеводах составляет сейчас мощную высокоразвитую ветвь органической химии. Перестали быть проблемой синтезы моносахаридов, в последние годы успешно выполнены синтезы ряда сложных олигосахаридов, становится реальностью синтез некоторых полисахаридов. Эта область сейчас постепенно приближается к такому состоянию, когда многообразие и богатство ее теоретического и методического арсенала будут соответствовать сложности объекта исследования.