

Pract-I-2014
.pdfНеидеальные растворы в широкой области концентраций компонентов не подчиняются законам Рауля и Генри. Однако в узких
областях, прилегающих к чистым компонентам, закон Рауля выполняется для растворителя, а для растворенного вещества выполняется закон Генри.
III.6.4. Азеотропные смеси.
Второй закон Гиббса - Коновалова
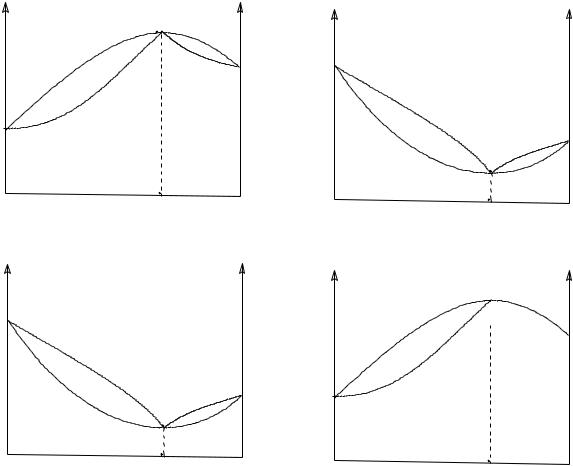
Иногда при отклонениях от закона Рауля на кривых давление - состав и температура - состав может появиться максимум или минимум в зависимости от того, положительные или отрицательные отклонения имеют растворы. В экстремальных точках кривая кипения жидкости обязательно соприкасается с кривой конденсации пара. Эта точка отвечает одинаковому составу равновесных жидкости и пара. Смеси, отвечающие экстремальным точкам, называются азеотропными, или нераздельно кипящими, или постоянно кипящими, так как при кипячении таких смесей образуется пар того же состава, что и исходная жидкость.
Второй закон Гиббса - Коновалова относится к системам, образующим максимумы или минимумы на кривых общего давления (для диаграмм в координатах давление - состав) или температур кипения смесей (для диаграмм в координатах температура - состав):
Экстремумы на кривых полного давления пара (или температур кипения) отвечают равновесию между раствором и насыщенным паром, при котором составы обеих фаз одинаковы, при этом максимум на кривой давления отвечает минимуму на кривой температуры и наоборот.
Образование азеотропной смеси (рис.III.9) в системе определяется двумя факторами: 1 - соотношением давлений пара (или соответственно температур кипения) компонентов в чистом состоянии и 2 - степенью отклонения системы от идеальности.
Если давление пара чистых компонентов системы мало отличаются друг от друга, то даже небольшое отклонение от идеальности может привести к образованию азеотропной точки, при этом азеотропная точка лежит обычно в области средних концентраций. При больших различиях в давлении пара компонентов требуется уже большее отклонение от идеальности для появления азеотропной точки, и положение ее смещено в область высоких концентраций одного из компонентов.
Азеотропная смесь для данных компонентов (при р или Т = const) однозначно определяется параметрами системы, что может быть подтверждено правилом фаз. В азеотропной точке имеются в равновесии две фазы - жидкость и пар, составы которых одинаковы Cж=Сп,
31
следовательно, появляется дополнительное уравнение, связывающее концентрации компонентов, которое должно быть учтено при подсчете числа степеней свободы системы, т.е. его следует вычесть. Тогда получим:
V = ( 2 - 1 ) + 1 - 2 = 0. Таким образом, система в азеотропной точке нонвариантна, а это значит, что ни давление, ни состав не могут быть выбраны произвольно.
p |
T=const |
M |
|
|
p |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
po |
A |
|
T=const |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
poB |
|
|
|
poB |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
M |
poA |
|
|
|
|
|
|
|
|
|
B |
|
xM |
А |
|
B |
|
xM |
А |
|
|
|
|
|
|
|||
T |
p=const |
|
|
|
T |
p=const |
|
|
|
|
|
|
|
M |
|
||
|
|
|
|
|
|
|
|
TB 
 TA
TA
TB
M TA
B |
xM |
А |
B |
xM |
А |
|
|
|
|||
|
а |
|
|
б |
|
Рис.III.9 Диаграммы кипения с образованием азеотропных смесей при отклонении от закона Рауля положительном (а) и отрицательном (б)
Несмотря на то, что азеотропная смесь при заданных условиях имеет строго определенный состав, она не является химическим соединением компонентов, образующих систему. Состав азеотропной смеси можно изменять, если проводить построение системы с азеотропной точкой в координатах давление - состав при Т =const, меняя значения температур,
32
что равносильно построению нескольких сечений в трехмерной диаграмме (см. рис.III.1). При этом можно наблюдать, что положение азеотропной точки изменится и по координате “давление” и по координате “состав”. Сказанное справедливо и для диаграмм, построенных в координатах температура - состав при р = const, если менять давление.
IV. ЭЛЕКТРОХИМИЯ РАСТВОРОВ ЭЛЕКТРОЛИТОВ
IV.1. ОСНОВНЫЕ ПОЛОЖЕНИЯ
Электрохимия - наука, изучающая превращения энергии химической реакции в электрическую, и наоборот. В электрохимии изучаются также физико-химические свойства растворов электролитов, процессы и явления на границах раздела фаз с участием заряженных частиц - ионов.
Электрохимические процессы имеют большое практическое значение в металлургии, в химической промышленности, в технологии гальванопластики, в современных научных методах исследования растворов электролитов и расплавов, а также методах анализа. Важную роль электрохимия играет в изучении процессов коррозии и разработке методов защиты металлов. Современная электрохимия активно занимается теорией разработки и создания топливных элементов и электрохимических преобразователей информации.
IV.1. ЭЛЕКТРОПРОВОДНОСТЬ РАСТВОРОВ СЛАБЫХ ЭЛЕКТРОЛИТОВ
Многие химические соединения в растворах, а также в расплавах частично или полностью распадаются на ионы. Такие соединения называются электролитами. Их растворы обладают ионной проводимостью. Процесс распада вещества на ионы называется
электролитической диссоциацией. Величина и знак заряда иона, а также количество ионов, образующихся при диссоциации из одной молекулы электролита, зависят от природы веществ. Основной количественной характеристикой электролитической диссоциации является степень диссоциации , которая показывает долю молекул, распавшихся на ионы, и принимает значения 0 1. Степень диссоциации при данной температуре зависит от природы вещества и растворителя, концентрации раствора, присутствия в растворе других электролитов и пр. Для слабых электролитов в разбавленных растворах 1.
33

Количественной характеристикой электролита может служить также
константа диссоциации Кс. |
|
|
|
|
|
|
|
|
|
КА |
|
|
|
|
К+ + А- |
|
|||
|
|
|
|||||||
|
|
|
|
|
|||||
КС |
|
СК С А |
,. |
(IV.1), |
|||||
СКА |
|||||||||
|
|
|
|
||||||
где С [моль/л] - концентрация электролита. Для бинарного электролита связь константы диссоциации и степени диссоциации
выражается соотношением
КА 
 К+ + А- С(1- ) С С
К+ + А- С(1- ) С С
KC |
2 С |
|
1 , |
(IV.2) |
Уравнение (IV.2) называется законом разбавления Оствальда.
Вместо концентрации можно использовать обратную ей величину - V - разведение, т.е. величину объема раствора, в котором содержится единица количества вещества: V=1/С [л/моль] (или [м3/моль] в единицах СИ). Константу диссоциации в этом случае выражают соотношением
KC |
|
|
2 |
. |
(IV.3) |
|
)V |
||||
|
(1 |
|
|
||
Константа и степень диссоциации могут быть определены из опытных данных электропроводности растворов. Электропроводность - это способность вещества проводить электрический ток - является величиной обратной электрическому сопротивлению
=1/R |
или |
1 |
|
S |
|
|
S |
, |
|
|
(IV.4) |
|
|
0 l |
|
|
|||||||
|
|
l |
|
|
|
|
|||||
где - электропроводность раствора (Ом-1), |
|
- |
удельное |
||||||||
сопротивление (Ом.м), S |
- сечение проводника (м2) |
и |
l |
- длина |
|||||||
проводника (м), 0 - удельная электропроводность раствора (Ом-1м-1).
Удельная электропроводность ( о) соответствует проводимости
слоя электролита, заключенного между двумя параллельными
электродами площадью 1 м2 на расстоянии |
1 м при градиенте |
потенциала 1В/м |
|
0=1/ ,.(Ом-1м-1). |
(IV.5) |
Удельная электропроводность является, по сути, измеряемой величиной (через измерение сопротивления раствора). Однако удельная электропроводность не дает полного представления об общих закономерностях в растворах электролитов. Для установления таких закономерностей лучше подходит введенная Р.Э. Ленцем мольная электропроводность.
34
Мольная электропроводность ( ) определяется как проводи-
мость слоя раствора электролита толщиной в 1 м, помещенного между параллельными электродами такой площади, чтобы заключенный между ними раствор содержал 1 моль вещества. Связь между удельной и мольной электропроводностями выражается соотношениями:
= о/С, |
(IV.6) |
или = оV, |
(IV.6a), |
если С (концентрация) имеет размерность моль/м3, а V - м3/моль. Мольная электропроводность в единицах СИ имеет размерность [Ом-1 м 2 моль-1 или См м2 моль-1 ], если же С - концентрация выражена в моль/л, то
|
01000 |
(IV.7) |
||||
|
|
С |
||||
|
|
|
|
|
||
Степень диссоциации связана с электропроводностью и |
|
|||||
выражается уравнением Аррениуса |
|
|
|
|
|
|
|
C |
., |
(IV.8) |
|||
|
|
|||||
|
|
|
||||
|
|
|
|
|
||
где С - мольная электропроводность при данной концентрации, а- мольная электропроводность при бесконечном разведении. Величина
представляет собой предельное значение с в условиях, когда С 0 и
1.
Г.Кольрауш, изучая разбавленные растворы сильных электролитов, установил правило аддитивности, согласно которому предельные значения подвижности ионов при бесконечном разведении не зависят от природы других ионов, присутствующих в растворе, т.е. электрическая подвижность или абсолютная скорость иона является его индивидуальной характеристикой. Абсолютная скорость иона - это путь, пройденный ионом в единицу времени при градиенте потенциала равной 1 Вм-1.
Таким образом, скорость иона ui = ui/ м2В-1с-1. Для химических процессов более важной индивидуальной характеристикой иона является
ионная электропроводность i = Fui или |
|
|
+=Fu+ |
и -=Fu- |
(IV.9), |
где F - число или постоянная Фарадея, равная 96484 Кл моль-1 |
|
|
Согласно правилу Г.Кольрауша о независимости движения ионов в
бесконечно разбавленных растворах для |
1,1-валентных электролитов |
|
можно получить соотношения: |
|
|
= + + - = F(u+ + u-) |
(IV.10). |
|
Это соотношение носит название закона Г.Кольрауша и при |
||
бесконечном разведении справедливо |
для сильных |
и слабых |
1,1-валентных электролитов. |
|
|
35
Объединив уравнения (IV.6) и (IV.8) с учетом закона Г.Кольрауша получим соотношение
|
C |
|
0 / C |
(IV.11), |
|||||
|
|
|
|
|
|
||||
|
|
|
|||||||
|
|
|
|
|
|
||||
которое лежит в основе расчетов констант диссоциации слабых кислот и произведений растворимости мало растворимых солей.
IV.2. ОСОБЕННОСТИ РАСТВОРОВ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ
Сильные электролиты в водных растворах практически полностью диссоциируют на ионы, концентрации ионов велики, электростатическое взаимодействие между ионами достаточно сильное, поэтому идеальные законы в таких случаях не применимы.
Чтобы сохранить простую форму термодинамических уравнений, описывающих свойства равновесных систем, Льюисом была введена новая функция, заменяющая концентрацию, названная активностью:
а = m, |
(IV.12) |
где а - активность ионов в растворе,- коэффициент активности ионов,
m - моляльность раствора электролита.
Коэффициент активности является мерой отклонения реальных систем от идеальных. Коэффициент активности «поправляет» концентрацию так, чтобы уравнения, полученные для идеальных систем, были справедливы и для реальных. В очень сильно разбавленных растворах взаимодействие между ионами незначительно и активность почти равна концентрации, а коэффициент активности стремится к единице.
Из опытных данных невозможно определить ни активность, ни коэффициент активности отдельных ионов, поэтому вводятся понятия
средней активности и среднего коэффициента активности.
Если реакция диссоциации электролита проходит по уравнению:
|
|
|
КqAn = qKn+ |
+ nAq-, |
|
|
||||||
то |
|
|
а |
q |
a |
n 1 / (q n) |
|
q |
a |
n |
1 / q n |
, |
а |
К |
|
a |
|
|
|
||||||
|
|
|
|
|
A |
|
|
|
|
|||
|
|
|
|
q |
1 / q n |
|
q |
|
1 / q n |
|
|
|
|
n |
|
n |
|||
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
K A |
|
|
|
|
||
(IV.13)
(IV.14)
(IV.15)
36

где а ,, - средняя активность и средний коэффициент активности иона в растворе.
Зная средний коэффициент активности, можно рассчитать
активность каждого иона: |
|
|
аК = a+ = mК |
аА=a- = mА, |
(IV.16) |
где mi - моляльность иона в растворе электролита определяется в соответствии с реакцией диссоциации электролита.
В случае если q = n, выражения для среднего коэффициента активности, средней активности и средней концентрации примут вид:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
, |
a |
a a |
|
. |
m |
|
m m m |
(IV.17), |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
т.е. для электролита типа КА, полностью диссоциированного |
|
|||||||||||||
|
|
|
|
|
|
mК = mА = m; |
|
|
m = m, |
|
||||
т.е. средняя моляльность ионов совпадает с моляльностью электролита.
По теории Льюиса коэффициенты активности зависят от заряда иона и ионной силы раствора, но слабо зависят от природы иона. Это значит, что для различных ионов с одинаковыми зарядами при равной ионной силе раствора коэффициенты активности примерно одинаковы.
Ионная сила раствора I характеризует суммарное воздействие электрических зарядов всех ионов на любой ион и равна полусумме произведений концентрации иона на квадрат его заряда для всех ионов,
находящихся в растворе |
|
|
||
I |
1 |
m z2 |
(IV.18) |
|
|
||||
2 |
|
i i |
|
|
Например, для раствора m(BaCl2) = 0,01 моль/кг, получим: |
||||
BaCl2 = Ва2+ + 2 Cl1- |
||||
mi = 0,01 |
0,01 |
2х0,01 |
||
m(Ва2+) = 0,01, m(Cl1-)= 0,02, zBa2+=2, zCl1- =1, поэтому
I = 1/2 (0,01 22 + 0,02 12 ) = 0,03.
А для смеси. m1(BaCl2) = 0,01 моль/кг и m2(AlCl3) = 0,003 моль/кг
ионная сила будет равна
I = 1/2 (0,01х22 + 0,02х12 + 0,003х32 + 0,009х12) = 0,048
Теория сильных электролитов, развитая П.Дебаем и Э.Хюккелем, приводит к следующему уравнению, связывающему средний коэффициент активности с ионной силой раствора (предельный закон Дебая-
Хюккеля):
lg A |
|
|
|
|
|
z z |
|
I , |
(IV.19) |
||
37
где А - коэффициент, зависящий от свойств растворителя, для водных растворов А = 0,509 (л/моль)1/2,
- средний коэффициент активности ионов, zi - заряд данного иона.
Предельный закон Дебая-Хюккеля строго выполняется для разбавленных растворов электролитов, при значениях I 0,02 и приближенно - для I 0,1 - 0,2, что позволяет рассчитывать коэффициенты активности для ионов разных зарядов в зависимости от ионной силы раствора.
IV.3. ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКИХ ПРОЦЕССОВ
Гальванический элемент - это система, в которой осуществляется прямое превращение энергии химических реакций в электрическую.
Гальванический элемент состоит из двух или более полуэлементов, каждый из которых представляет собой электрод с электронной проводимостью, погруженный в раствор электролита с одноименными ионами. Растворы электролитов двух полуэлементов обычно соединяют друг с другом электролитическим солевым мостиком, который обеспечивает электрический контакт между растворами, но препятствует их взаимной диффузии. После соединения электродов металлическим проводником в цепи возникает электрический ток и может быть получена электрическая работа. Возникновение электрического тока связано в основном с изменением энергии Гиббса химической реакции, протекающей в гальваническом элементе.
При обратимом изотермическом процессе в такой системе совершается максимальная полезная работа (Wм.п), равная изменению энергии Гиббса химической реакции. Электрическая работа (Wэл) является одним из видов полезной работы, тогда в отсутствии других видов работы, можно записать
Wм.п = G = Wэл = z F E, |
(IV.20) |
||
|
E |
G |
|
или |
zF |
(IV.21). |
|
|
|
|
|
где Е - электродвижущая сила, В,
F -число Фарадея или постоянная Фарадея, 96484 Кл/моль
z - число электронов, участвующих в электрохимической реакции. Поскольку для G нам известно несколько выражений, то в
дальнейшем мы используем их для получения различных выражений и для ЭДС.
38
Электродвижущая сила (ЭДС) возникает в системе в результате протекания окислительно-восстановительной электрохимической реакции и является алгебраической суммой скачков потенциалов, возникающих
на границах раздела фаз: |
|
|
|
Е = 1 + 2 + дифф. |
(IV.22) |
где 1 и |
2 - электродные потенциалы, возникающие на границе |
|
электродраствор, |
|
|
дифф. - диффузионный потенциал, возникающий на границе растворраствор. Его обычно убирают с помощью электролитического солевого
мостика, т.е. дифф = 0,. |
|
Таким образом, Е = i . |
(IV.23) |
Напряжение в электрохимической цепи достигает максимального значения при установлении электрохимического равновесия на всех границах раздела фаз. Измерение этого напряжения в системе проводится, по А.Вольта, при правильно разомкнутой цепи, т.е. при компенсации этого напряжения разностью потенциалов от внешнего источника тока. Это напряжение и называется ЭДС.
Что такое электродный потенциал и как он возникает? Электродный потенциал возникает при погружении металла в раствор своей хорошо растворимой соли. При взаимодействии металла с дипольными молекулами воды происходит переход ионов металла в раствор. При этом металл заряжается отрицательно, а раствор - положительно. Устанавливается равновесие, при этом ионы металла остаются вблизи электрода и т.о. создается двойной электрический слой с определенным зарядом (подобно конденсатору) - это и есть электродный скачок потенциала. В электрохимии принято реакции, протекающие на
электродах записывать в сторону восстановления |
|
МZ+ + ze M |
(IV.24) |
Равновесие реакции можно охарактеризовать изотермой реакции:
G Go RT ln |
a M |
|
a |
|
|
|
M Z |
где аМze - активность катиона в растворе, аМ электроде = 1, тогда c учетом уравнения (IV.20)
(IV.25),
- активность металла на
|
G |
|
|
Go |
RT |
|
|
1 |
|
|||
|
|
|
|
|
|
|
|
ln |
|
|
(IV.26) |
|
zF |
|
zF |
zF |
a |
M Ze |
|||||||
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
или о |
RT |
ln a |
|
|
|
|
|
|
(IV.27) |
|||
|
|
Ze |
|
|
|
|
||||||
|
|
ZF |
|
M |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Уравнение (IV.27) представляет собой уравнение электродного потенциала Нернста, где:
39
- потенциал электрода при активности ионов в растворе равной
a M ze a ,
о - стандартный потенциал электрода при а+= 1.
Электроды по типу протекающей на них электрохимической реакции условно делятся на три рода.
Электроды первого рода - это электроды обратимые относительно катиона. Схема электрода записывается: Mz+ M, а реакция на электроде Mz+ + ze = M соответствует выражению для электродного потенциала:
|
|
|
o |
|
|
RT |
ln a |
|
(IV.28). |
z |
|
z |
|
|
|||||
M |
/ M |
M |
/ M |
zF |
|
||||
|
|
|
|
|
|
||||
Большинство из них - это металлические электроды (См. табл.IV.1 №№ (электродов) 1-6,10, 16). К электродам первого рода относятся также водородный и амальгамные электроды.
|
|
|
Таблица IV.1 |
|
|
Стандартные электродные потенциалы при 25oС |
|||
|
|
|
|
|
№ |
Электрод |
Реакция на электроде |
|
0, В |
1 |
Zn2+Zn |
Zn2+ + 2e = Zn |
|
-0,763 |
2 |
Cr3+Cr |
Cr3+ + 3e = Cr |
|
-0,744 |
3 |
Fe2+Fe |
Fe2+ + 2e = Fe |
|
-0,440 |
4 |
Cd2+Cd |
Cd2+ + 2e = Cd |
|
-0,403 |
5 |
Pb2+Pb |
Pb2+ + 2e = Pb |
|
-0,126 |
6 |
H+H2, Pt |
H+ + 1e = 1/2 H2 |
|
0 |
7 |
Sn4+,Sn2+ Pt |
Sn4+ +2e = Sn2+ |
|
0,15 |
8 |
Cl-AgCl, Ag |
AgCl + 1e =Ag + Cl- |
|
0,222* |
9 |
Cl-Hg2Cl2, Hg |
1/2 Hg2Cl2 + 1e = Hg + Cl- |
|
0,268* |
10 |
Cu2+Cu |
Cu2+ + 2e = Cu |
|
0,337 |
11 |
Fe(CN)63-, |
Fe(CN)63- + 1e = Fe(CN)64 |
|
0,366 |
|
Fe(CN)64-Pt |
|
|
|
12 |
OH-O2, Pt |
1/4 O2 + 1/2 H2O + 1e = OH- |
|
0,401 |
13 |
I-I2, Pt |
I2 + 2e = 2 I- |
|
0,536 |
14 |
H+xr, Pt |
1/2 C6H4O2+H++1e = 1/2 C6H4(OH)2 |
|
0,700 |
15 |
Fe3+, Fe2+Pt |
Fe3+ + 1e = Fe2+ |
|
0,771 |
16 |
Ag+Ag |
Ag+ + 1e = Ag |
|
0,799 |
17 |
Cl-Cl2, Pt |
1/2 Cl2 + 1e = Cl- |
|
1,360 |
40