

И проводящей тугоухостью
синдром Ушера, встречающийся у 2,5% глухих (врожденная нейросенсорная глухота и пигментный ретинит). Типичны врожденная нейросенсорная патология слуха, отсутствие вестибулярных реакций и медленно прогрессирующий пигментный ретинит с началом на 1-2-м десятилетии жизни. Из других глазных симптомов наблюдаются катаракта, дегенерация сетчатки, иногда глаукома. В четверти случаев — умственная отсталость, иногда шизофрения.
Наследуется аутосомно-рецессивно (рис. 21)

Рис. 21. Синдром Ушера
пороки развития скелета и болезни соединительной ткани. Среди этой группы стойких нарушений слуха можно выделить: черепно-лицевой дизостоз, или синдром Крузона. Основными проявлениями синдрома Крузона являются деформации черепа (брахицефалия, оксицефалия), экзофтальм, мелкие орбиты, гипоплазия верхней челюсти (рис. 22). Наблюдаются также гипертелоризм, расходящееся косоглазие, нистагм, клювовидный нос, иногда расщелина твердого нёба или язычка, двусторонняя атрезия наружного слухового прохода, различные степени снижения слуха, интеллекта, зрения.

Рис. 22. Синдром Крузона
Нижнечелюстно-лицевой дизостоз, или синдром Тричера-Коллинза. Основными клиническими проявлениями синдрома Тричера — Коллинза являются двусторонняя гипоплазия скуловых костей и орбит, колобома нижних век, антимонголоидный разрез глазных щелей, отсутствие ресниц на нижнем веке, аномалии ушных раковин, проводящая глухота, гипоплазия нижней челюсти (рис. 23). Тип наследования — аутосомно-доминантный;

Рис. 23. Синдром Тричера-Коллинза
наследственный нефрит с глухотой, или синдром Альпорта. Заболевание проявляется различными нарушениями функции почек (гематурия, протеинурия и др.), переходящими часто в почечную недостаточность. В 50% случаев отмечаются нейросенсорные расстройства слуха, начинающиеся с первых лет жизни. У 15% больных выявляется катаракта или другие аномалии глаз. Предполагается генетическая гетерогенность синдрома (6 форм с различными клиническими особенностями и неодинаковыми типами наследования - аутосомно-доминантным, Х-сцепленным рецессивным, аутосомно-рецессивным). Синдром Альпорта встречается у 1% детей с врожденными нарушениями слуха;
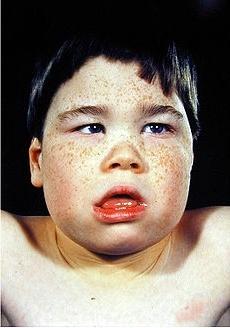
Рис. 24. Синдром Альпорта
Эндокринная патология. К этой группе заболеваний отнесены устойчивые сочетания наличия зоба с нейросенсорной глухотой. Заболевание носит название синдрома Пендреда, который встречается у 10% больных с врожденной глухотой. Для заболевания характерна врожденная нейросенсорная глухота. С 5-8-летнего возраста отмечается увеличение щитовидной железы за счет развития диффузного зоба. В некоторых случаях наблюдается умственная отсталость. Наследуется аутосомно-рецессивно (рис. 25).

Рис. 25. Синдром Пендреда
патология нервной системы, например, синдром атаксии, гипогонадизма, умственной отсталости и нейросенсорная глухота, носящий наименование синдрома Ричардса-Рандля. Для больных характерны задержка моторного развития, атаксия, недоразвитие вторичных половых признаков, деформации стоп, когтеобразная деформация кисти, кифосколиоз, атрофия мышц, умственная отсталость. Глухота носит прогрессирующий характер. Тип наследования синдрома — аутосомно-рецессивный.
