

Федеральное агентство по образованию
Федеральное государственное образовательное
учреждение высшего профессионального образования
«Южный федеральный университет»
Э.А.Бикяшев основы теории электролитической диссоциации
Учебное пособие
г. Ростов-на-Дону
2009
Бикяшев Э.А.
Основы теории электролитической диссоциации:
Учебно-методическое пособие. – Ростов-на-Дону, 2009 - 29 с.
Аннотация
Настоящее учебно-методическое пособие предназначено для обеспечения теоретических и практических занятий по курсу "Неорганическая химия" для студентов биолого-почвенного факультета ЮФУ.
Пособие содержит краткий анализ механизмов электролитической диссоциации в зависимости от структуры потенциального электролита, а также от способа воздействия на его связи (модуль 1). Показано, что для ионных соединений диссоциация наблюдается в процессе их плавления или растворения в полярных жидкостях. В то же время вещества молекулярного строения могут подвергаться электролитическому распаду только в среде ионизирующего растворителя. В модуле 2 рассматриваются количественные закономерности диссоциации в растворах. Обсуждается зависимость степени диссоциации от структуры электролитов и внешних факторов (параметров состояния).
Модуль №1
Электролитический распад связей
Механизм электролитической диссоциации
1.1 Основные понятия
Некоторые исторические вехи
Развитие представлений о механизмах диссоциации тесно связано с историей развития теории растворов и электрохимией растворов (закономерности, описывающие электропроводность растворов и процессы электролиза). Так, еще в 1805г. Ф.И.Гротгус высказал предположение, что в растворах при приложении электрического поля растворенное вещество распадается на противоположно заряженные частицы, которые затем нейтрализуются (разряжаются) на электродах. Позже М.Фарадей назвал подобные частицы ионами (в переводе с греческого "ион" – идущий, катион – "идущий к катоду" положительный ион, анион – "идущий к аноду" отрицательный ион), а вещества подвергающиеся электролизу, а, следовательно, и распаду на ионы под действием тока – электролитами. В 1878г. Р.Э.Ленц, изучая зависимость электропроводности растворов от приложенного напряжения, высказал мнение, что молекулы веществ уже при растворении могут распадаться на ионы. Эта гипотеза в дальнейшем нашла подтверждение в экспериментах С.Аррениуса по изучению электропроводности водных растворов кислот, оснований солей разной концентрации. Тот факт, что эквивалентная электропроводность1 таких растворов увеличивалась при разбавлении, стремясь к некоторому пределу, С.Аррениус объяснил не растущей подвижностью ионов, а увеличением их концентрации. Таким образом, уже в 1883г. была высказана точка зрения, что степень диссоциации веществ в водных растворах увеличивается при их разбавлении. Большое значение для дальнейшего развития теории диссоциации имела известная работа Вант-Гоффа «Химическое равновесие в системах газов и разбавленных растворов» (1885). В ней сообщалось, реальное понижение температуры замерзания растворов солей, кислот и оснований превышает рассчитанное теоретически по закону Рауля. Вант-Гофф установил, что эти отклонения носят систематический характер. Например:
|
Состав водного раствора |
|
|
|
|
i
=
|
|
Т = 298 К |
|||||
|
|
|
|
|
|
|
|
KCl (CM=0.2моль/л) |
0.673 |
0.372 |
896,88 |
495,5 |
1.81 |
|
|
|
|
|
|
|
|
MgCl2 (CM=0.1моль/л) |
0.519 |
0.186 |
694,24 |
247,76 |
2.79 |
|
|
|
|
|
|
|
 ,
°С
,
°С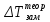 ,
°С
,
°С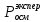 ,кПа
,кПа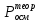 ,
кПа
,
кПа
Таким образом, было показано, что экспериментальные значения осмотического давления, понижения температуры замерзания растворов электролитов (а также других коллигативных свойств) могут быть согласованы с известными на тот момент формулами путем введения дополнительного поправочного множителя i, получившего название изотонический коэффициент.
Росм
= i·CM·R·T;
ΔTзам
= i·К·Сm;
ΔTкип
= i·Е·
Сm;
Рпара
= Ро·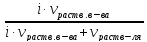 (1.1)
(1.1)
Для растворов многих (особенно органических: глицерин, сахароза) веществ изотонический коэффициент следует считать равным единице, его можно не учитывать в соответствующих формулах. Для растворов кислот (в т.ч. органических), оснований и солей i > 1, что равносильно самопроизвольному увеличению концентрации частиц по сравнению с той, что создавалась в процессе растворения.
В 1887 году появилась знаменитая статья Аррениуса «О диссоциации растворенных в воде веществ», в которой были приведены дополнительные данные по изотоническим коэффициентам. Было показано, что они увеличиваются при разбавлении растворов электролитов и стремятся при этом к некоторым целочисленным значениям, например, для KCl i→2, MgCl2 i→3. Обобщая эти данные с концентрационными зависимостями мольной электропроводности, С.Аррениус делает вывод о том, что это может быть объяснено только самопроизвольно идущими процессами распада растворенных веществ на ионы. Причем степень диссоциации увеличивается при разбавлении растворов. Предполагая, что мольная электропроводность λМ пропорциональна степени диссоциации
λМ = К·α, (1.2)
а также, сделав вывод на основе концентрационных зависимостей λМ и i, что при предельном разбавлении наблюдается полный распад на ионы (α = 1), С.Аррениус получает выражение для экспериментального определения степени электролитической диссоциации:
при бесконечном разбавлении λМ(∞) = К,
тогда λМ = λМ(∞) ·α и, соответственно,
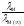
α = (1.3)
(ниже будут выведены несколько других важных формул).
Тем самым чисто качественная гипотеза превратилась в количественную теорию, которая могла быть проверена экспериментально. В 1888 году С.Аррениус предложил способ определения основности кислот по величине электропроводности их растворов и показал, что скорость химической реакции в растворах зависит только от диссоциированной части растворенного вещества (от концентрации ионов).
В том же году В.Оствальд вывел для бинарных слабых электролитов зависимость, которую назвал законом разбавления. В этом частном случае закона действующих масс сформулированы соотношения между константой диссоциации электролита, электропроводностью и концентрацией раствора. В дальнейшем этот закон подвергался неоднократной проверке. Было найдено, что для сильных электролитов и концентрированных растворов он неприменим. Как позже стало понятно, это было вызвано тем, что в теории С.Аррениуса не учитывалось взаимодействие между ионами и между ионами и растворителем.
Потребовались многочисленные исследования ученых конца XIX и начала XX века (Н. Бьеррум, П. Дебай и Э. Хюккель), чтобы объяснить, что необычное поведение сильных электролитов связано с очень высокой концентрацией ионов в их растворах и очень сильными кулоновскими силами притяжения (Гл.2.4).
В 1888 году В.Ф.Нернст, сравнив скорость диффузии ионов со скоростью движения ионов при электролизе, показал, что эти числа совпадают. В 1889 году на основе теории осмотического давления и теории электролитической диссоциации Нернст разработал теорию возникновения гальванического тока, обосновал возникновение двойных электрических слоев в приэлектродных участках раствора.
В 1889–1892гг. И.А.Каблуков и В.А.Кистяковский ввели понятие о сольватации ионов и, тем самым объединили теорию диссоциации С.Аррениуса с химической теорией растворов Д.И.Менделеева.
Дальнейшие существенные дополнения и корректировки теории диссоциации были продиктованы развитием представлений о строении атомов и веществ.