

12.7. Згортальна та фібринолітична системи крові
Згортання крові (гемокоагуляція) – життєво важливий захисний фізіологічно-біохімічний процес, спрямований на зменшення крововтрати при порушенні цілісності кровоносної системи, який забезпечується функціонуванням системи гемостазу.
Система гемостазу є однією з найважливіших у життєдіяльності організму, оскільки вона забезпечує структурну цілісність судинної стінки в нормі та при її пошкодженні; підтримує замкнутість кров’яного русла та попереджує кровотечі; підтримує стабільний агрегатний стан крові – створює оптимальні умови для кровоплину і обмінних процесів; запобігає внутрішньосудинному згортанню крові; виконує роль ідеального природного клею та "консерванта" пошкоджених тканин; забезпечує лізис тромбів з відновленням кровоплину та створенням передумов для оптимальної регенерації пошкоджених тканин.
Функціонування системи гемостазу забезпечують структурні елементи судинної стінки (ендотеліальні клітини, а саме їх базальні мембрани. які відіграють роль бар’єра між кров’ю та судинною стінкою і забезпечують необхідний обмін між ними), клітинні елементи крові (тромбоцити, базофільні гранулоцити) та фактори, що містяться у плазмі крові (їх налічують понад 40). Останні можна розподілити на дві групи: коагулянти, які забезпечують процес згортання крові, і антикоагулянти – інгібітори коагуляції.
Із фізіологічної точки зору розрізняють судинно-тромбоцитарний або первинний коагуляційний гемостаз (забезпечує зупинку кровотечі з мікросудини з утворенням «білого» тромба) і вторинний коагуляційний (формування та закріплення фібринового тромба).
Ключову роль у формуванні судинно-тромбоцитарного гемостазу відіграють тромбоцити – кров'яні пластинки, поліморфні без'ядерні утвори, оточені мембраною. Їх кількість у крові дорослої людини становить 150 – 300 109 л. У загальному об'ємі циркулюючої крові міститься 60-75 % усіх тромбоцитів, які синтезує червоний кістковий мозок, решта – в селезінці. Важливу роль відіграє мембрана тромбоцитів, що утворена глікокаліксом, фосфоліпідами та глікопротеїнами, які забезпечують адгезію та агрегацію цих клітин.
Тромбоцити містять пластинчасті фактори, які беруть участь у первинному гемостазі. Не зважаючи на відсутність ядра, вони здатні виконувати всі функції клітини, крім синтезу ДНК.
У тромбоцитах наявні два види гранул: щільні, у яких упаковані небілкові речовини, що виділяються під час активації пластинок (АДФ, АТФ, катехоламіни й серотонін, кальцій), та α-гранули лізосомальної природи, які відповідають за виділення білків іншого складу, ніж гідролази у хромосомах. Це антигепариновий фактор 4, b–тромбоглобулін, фактор Віллебранда, тромбосподин, фібронектин і тромбоцитарний фактор росту (PDGF), який також виділяється макрофагами та ендотеліоцитами, відіграє важливу роль в епітелізації рани і є потужним мітогеном для міоцитів кровоносних судин.
Основними фізіологічними властивостями тромбоцитів є унікальна здатність до адгезії та агрегації; секреція (реакція вивільнення) адреналіну, норадреналіну, серотоніну, які підтримують спазм ушкоджених судин; пристінкове розташування в судинах, яке зменшує проникність капілярів і забезпечує ангіотрофічну функцію; здатність виділяти гемостатичні фактори (тромбопластична функція).
12.7.1. Судинно-тромбоцитарний гемостаз – тромбоцитарна реакція, тобто реакція тромбоцитів на порушення цілісності стінки судин, яка формується паралельно до реакції самих судин на пошкодження - їх скорочення в місці пошкодження, шунтування крові вище пошкодженої ділянки. За рахунок судинно-тромбоцитарного гемостазу може самостійно припинитись кровотеча із невеликих судин, але при пошкодженні великих судин цього механізму недостатньо. Тут він виступає лише первинним гемостазом, з якого починаються всі фази зупинки кровотеч. Після пошкодження судин послідовно запускаються наступні етапи:
1. Рефлекторний спазм судин починається зразу після пошкодження. Він обумовлений місцевими рефлекторними механізмами і підтримується реакцією гладких м'язів судин пошкодженої ділянки на вазоактивні сполуки, які утворюються тут. Крім цього, при послідовному руйнуванні із тромбоцитів виділяються речовини, які звужують судини (серотонін, адреналін, тромбоксан). Спазм судин розвивається досить швидко, але через декілька хвилин може припинитись і кровотеча відновиться. Тому для зупинки кровотечі необхідно, щоб підключились інші механізми гемостазу.
2. Адгезія - приклеювання тромбоцитів до місця пошкодження. В ініціації цього процесу важлива роль належить волокнам колагену, до яких прилипають негативно заряджені тромбоцити. При цьому тромбоцит змінює свою форму і викидає довгі ниткоподібні відростки псевдоподії (рецептори). Найважливішим плазменним фактором адгезії тромбоцитів є фактор Віллебранда - глікопротеїн, який синтезується ендотелієм судин (він накопичується також у тромбоцитах). Отже ефективність адгезії обумовлена взаємодією трьох компонентів:
1) специфічних глікопротеїнів - рецепторів мембран тромбоцитів;
2) колагену;
3) фактора Віллебранда й деяких інших білків – тромбоспондину, фібронектину. Останній утворює своєрідні містки між коллагеном субендотелію судин і рецепторами тромбоцитів.
Глікопротеїн I складається із двох дисульфіднозвязаних субодиниць, Iа й Ib. Глікопротеїн Iа, або глікокаліцин (маса молекули 130 000 – 160 000) є рецептором для фактора Віллєбранда й необхідний для повноцінної адгезії тромбоцита до колагену. Глікопротеїн 1b (маса молекули 22 000) забезпечує рецептор для тромбіну і тому без нього немає повноцінної тромбін-агрегації. Глікопротеїн II також складається із двох субодиниць, IIa й IIb. Він необхідний для агрегації тромбоцитів всіх видів. Глікопротеїн III (маса молекули 114 000) розглядають як варіант глікопротеїна II. Тому його позначають глікопротеїном IIIa або IIb. Глікопротеїн IV (синонім глікопротеїн IIIb, маса молекули 85 000 – 100 000) відрізняється від інших глікопротеїнів резистентністю до трипсину й хімотрипсину. Глікопротеїн V (маса молекули 68 000 – 89 000) селективно гідролізується тромбіном, необхідний для здійснення тромбіну-агрегації. Тромбін, взаємодіючи із глікопротеїном V, формує на тромбоциті рецептори до активованих факторів X й V, які, приєднавшись до тромбоцита, уже не нейтралізуються антитромбіном III і гепарином. У свою чергу, реалізується локальне згортання крові в зоні тромбування судини.
Крім глікопротеїнових рецепторів на тромбоцитарній мембрані є рецептори до агонистів: тромбіну, АДФ, фібриногену, колагену, простагландинів, серотоніну, Fc-фрагменту імуноглобуліну, компонентів комплементу, інсуліну. На тромбоциті є й адренорецептори.
Ф
Рис. 12.7. Адгезія
тромбоцитів до
колагенових волокон
ендотелію
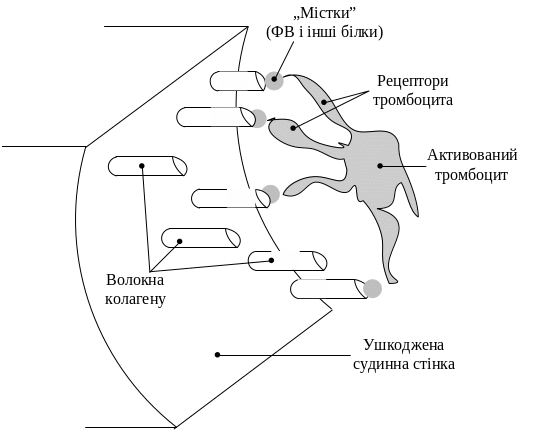
3. Агрегація (скупчення) тромбоцитів. Поява ниткоподібних відростків, зміна форми тромбоцитів відбуваються ще під час підходу до місця пошкодження. Це сприяє «склеюванню» тромбоцитів між собою (по 10 - 20) і прилипанню у такому стані до стінки судини. Внаслідок цього формується первинний, так званий «білий» тромб, який прикриває пошкоджену ділянку. Але він ще не щільний і може пропускати плазму крові.
Д
Рис.
12.8.
Участь тромбоцитів в первинному
гемостазі
шляхом вивільнення з гранул біологічно
активних речовин

В
Рис.
12.9. Шляхи
метаболізму арахідонової кислоти в
тромбоцитах і ендотеліоцитах і
місця його блокування:
1 - інгібітори циклооксигенази (аспірин,
сульфопіразол та інші протизапальні
нестероїдні препарати) 2 - інгібітори
тромбоксансинтази (похідні імідазолу)
3 - комбіновані інгібітори
тромбоксансинтази
та блокатори рецепторів для тромбоксану
А2
і ендопероксидів (рідогрел, пікотамід)

Тромбоксан А2 вивільняє із гранул клітин депоновані іони кальцію й АДФ, які виштовхуються на поверхню клітини, де із глікопротеїном IIb/IIIa вони формують рецептори до фібриногену, молекули якого створюють стяжки між клітинами, утримують їх у конгломераті й укрупнюють за рахунок викиду нових порцій АДФ із втягуванням у процес інтактних кров'яних пластинок.
Цей процес у кровотоку був би необмеженим, якби із зони неушкодженого ендотелію у відповідь на виділення з агрегуючих тромбоцитів АДФ і тромбоцитарного фактора ІІІ, що стимулює “внутрішній шлях” утворення тромбіну, не включався б арахідоновий каскад в ендотеліоцитах, що призводить до утворення ПГI2, що перешкоджає подальшій агрегації тромбоцитів, чим й обмежує розмір тромбоцитарного вала ("тромбоцитарного корка") межами ушкодженої ділянки судини.
4. Ретракція «білого» тромба. Із зруйнованих тромбоцитів вивільняється пластинчастий фактор (ПФ-6) – тромбостенін. Його здатність до скорочення обумовлює зменшення розміру пошкодженої ділянки та ущільнення згустка, утворення якого відбувається впродовж 1-3 хв з моменту ушкодження.
12.7.1.1. Порушення судинно-тромбоцитарного гемостазу. У зв’язку з природженими або набутими дефектами тих чи інших складових судинно-тромбоцитарного гемостазу може виникати низка патологій, які будуть у подальшому перешкоджати перебігу коагуляційного гемостазу, тобто, утворенню так званого «червоного тромба» (табл. 12.2).
Таблиця 12.2. Найважливіші фактори судинно-тромбоцитарного гемостазу та клінічні прояви їх дефіциту
Фактори |
Місце утворення, локалізація |
На які процеси впливають |
Синдроми дефіциту |
Глікопротеїн Іb |
Мембрана тромбоцита (рецептор) |
Адгезія |
Хвороба Бернара-Сульє |
Глікопротеїни ІІ b та ІІІ a |
Мембрана тромбоцита (рецептор) |
Незворотна агрегація |
Тромбастенія Гланцмана-Негелі |
Фактор Віллебранда |
Субендотелій ушкоджених судин, плазма |
Адгезія |
Хвороба Віллебранда |
Тромбоксан А2 |
Тромбоцити |
Незворотна агрегація, спазм судин |
Природжені та набуті тромбоцитопенії |
Простациклін |
Судинна стінка |
Інгібітор агрегації тромбоцитів |
Надмірна схильність тромбоцитів до агрегації та утворення тромбів |
Велике значення в останні роки надають існуванню тромбоцитопатій з порушенням реакції вивільнення, до яких, очевидно, належить більша частина “непояснених” форм кровоточивості. В основі цих захворювань лежить порушення реакції вивільнення із гранул кров'яних пластинок біологічно активних речовин, необхідних для підтримання нормальної агрегації тромбоцитів (АДФ, адреналіну, серотоніну, тромбоксану А2 тощо). Існують природжені й набуті форми такої патології. Набуті порушення реакції вивільнення часто обумовлені впливом на кров'яні пластинки лікарських речовин (ацетилсаліцилова кислота, індометацин, бутадіон, ібупрофен, імідазол, дипіридамол (курантил), реополіглюкін, папаверин, еуфілін, ізоптин (верапаміл), корінфар, аміназин, амітриптилін, карбеніцилін, надмірні дози пеніциліну тощо). При цьому в багатьох випадках під дією лікарських препаратів виникає зниження активності циклооксигенази або тромбоксансинтетази, тим самим пригнічується процес утворення тромбоксану А2, необхідного для повноцінної агрегації тромбоцитів. Наприклад, ацетилсаліцилова кислота навіть при однократному прийомі в дозі 0,65 г і більше блокує реакцію вивільнення, знижуючи адгезивность тромбоцитів, другу хвилю агрегації, подовжуючи час кровотечі тощо. Така дія ацетилсаліцилової кислоти на тромбоцити стабільна й ліквідується лише через 4 – 6 днів після прийому препарату, тобто коли більша частина ушкоджених тромбоцитів заміщається знову утвореними в кістковому мозку.
Подібними властивостями володіє ще одна група тромбоцитопатій – так звані хвороби недостатнього пулу нагромадження або зберігання. У цю групу входять спадкові тромбоцитопатії, для яких характерна нездатність накопичувати й, відповідно, виділяти необхідні для агрегації біологічно активні речовини (АДФ, серотонін, адреналін тощо).
12.7.2. Коагуляційний гемостаз. Одночасно з первинним (судинно-тромбоцитарним) гемостазом розвивається вторинний (коагуляційний), який забезпечує зупинку кровотечі із тих судин, для яких було недостатньо попереднього етапу. Тромбоцитарний згусток не витримує високого тиску крові і при зменшенні реакції рефлекторного спазму може вимиватися. Тому на зміну йому формується червоний тромб. Основою його утворення є перетворення розчинного фібриногену на нерозчинний фібрин з формуванням сітки, в яку захоплюються формені елементи крові (утому числі еритроцити, які надають тромбу характерного червоного забарвлення). Фібрин утворюється під впливом фермента тромбіну, який за умов норми відсутній у крові. В ній міститься його попередник в неактивній формі - протромбін, для активації якого необхідний фермент протромбіназа. Процес утворення активної протромбінази складний, вимагає взаємодії багатьох факторів плазми, клітин, тканин і триває 5 - 7 хв.
12.7.2.1. Фактори згортання крові. Всі фактори згортання присутні в плазмі в неактивній формі. Вони позначаються римськими цифрами й відповідними назвами, у яких відображена їх функція (наприклад, фактор XI - плазменний попередник тромбопластину), прізвища хворих із уперше виявленим у них дефіцитом того або іншого фактора (фактор XII - фактор Хагемана, фактор Х - фактор Стюарта-Прауэра тощо) або прізвища авторів, що описали даний фактор вперше (наприклад, фактор Віллєбранда) (табл. 12.3). Для позначення активованих факторів згортання додається буква “а”. Варто пам'ятати також, що фактор VI вилучений із класифікації, тому що являє собою активований фактор V. Деякі з факторів згортання не мають цифрових позначень.
Таблиця 12.3. Фактори згортання крові
Назва |
Характеристика |
I, Фібриноген |
Глікопротеїн з молекулярною масою 340 000 Дальтон, складається з 2 946 послідовних амінокислот, і являє собою димер, у кожній одиниці якого містяться три поліпептидні ланцюги, з'єднані дисульфідними містками. Фактор I у тому вигляді, у якому він виробляється паренхіматозними клітками печінки й надходить у кров, називається фібриногеном А, на відміну від фібриногену В, що осаджується із плазми вітаміном К. Під дією тромбіну фібриноген перетворюється на нерозчинний у крові фібрилярний білок — фібрин |
II, Протромбін
|
належить до еуглобулінів, cинтезуєтся в печінці при участі вітаміну К. Під дією протромбінази утворюються -, - і -тромбіни. -тромбін має потужну згортальну активність відносно фібриногену, але швидко нейтралізується антитромбіном III. -тромбін резистентний до гепарину й антитромбіну III. -тромбін не має згортальної активності, але йому властива естеразна й фібринолітична дія. |
III, Тканинний тромбопластин |
Ліпопротеїн, бере участь в зовнішньому шляху активації фактора X. З формених елементів тканинний тромбопластин можуть синтезувати тільки моноцити. |
IV, Іони Кальцію
|
Мають першорядне значення для активації протромбінази й перетворення протромбіну на тромбін. Прискорюють утворення фібрину. Необхідні для взаємодії факторів згортання з фосфоліпідною поверхнею клітин. Кальцій здатний зв'язувати гепарин, завдяки чому згортання крові прискорюється. Без кальцію порушується агрегація тромбоцитів і ретракція кров'яного згустку. Іони кальцію інгібуть фібриноліз. |
V, Проакселерин (Ас-глобулін плазми) |
β-глобулін, синтезується в печінці, не залежить від вітаміну К. перетворюється автокаталітично за наявності тромбіну. Необхідний для утворення внутрішньої протромбінази і для перетворення протромбіну на тромбін, коли в комплекс включаються фактор Ха, Са2+і фосфоліпіди. Під час згортання крові фактор повністю використовується, як і фактор II, тому в сироватці не виявляється. |
VI, Акселерин (Ас-глобулін сироватки) |
β-глобулін, активна форма фактора V. |
VII, Проконвертин
|
Серинова протеїназа з аргінін-естеразною активністю, бере участь в активації фактора X, синтезується в печінці за участі вітаміну К. Фактори ХII, Ха, каллікреїн можуть перетворювати фактор VII на VIIa. Сприяє утворенню тканинної протромбінази й перетворенню протромбіну на тромбін. Фактор VII у циркулюючій крові активує фактор X. Ця дія підсилюється після активації проконвертину тканинним тромбопластином. |
VIII, Антигемофільний глобулін А
|
Складний глікопротеїн, синтезується в печінці, селезінці, клітинах ендотелію, лейкоцитах, нирках. У крові циркулює у вигляді комплексу із трьох субодиниць, позначуваних VIIIK(коагулююча одиниця), VIII-АГ (основний антигенний маркер) і VIII-фВ (фактор Віллебранда, пов'язаний з VIII-АГ). VIII-фВ регулює синтез коагулянтної частини антигемофільного глобуліну—VIIIK. |
IX, Антигемофільний глобулін В (автопротромбін, фактор Крістмаса) |
Серинова протеїназа, бере участь в утворенні активного тромбопластину; синтезується в печінці за участі вітаміну К. У процесі згортання крові не використовується повністю й залишається в сироватці в більш активному стані, ніж у плазмі. |
X, Тромботропін (автопротромбін С, фактор Стюарта - Прауера)
|
Глікопротеїн з масою молекули 54 200-56 000 Да, складається із двох поліпептидних ланцюгів: тяжкого (з молекулярною масою 38 000), на якому перебуває активний центр, і легкого — із залишком карбоксиглютамінової кислоти, необхідної для приєднання до фосфоліпідів.. Серинова протеїназа, синтезується в печінці за участі вітаміну К бере участь в утворенні тромбіну. Активується трипсином і ферментом з отрути гадюки Рассела. |
XI, Попередник плазмового тромбопластину (фактор Розенталя) |
Глікопротеїн з масою молекули 160 000 Да. У процесі згортання крові повністю не використовується, тому виявляється у великій кількості в сироватці. Активна форма утворюється за участі факторів ХIIа, Флетчера й Фітцжеральда-Фложе. |
XII, Фактор контактної активації (Хагемана)
|
Сполука з масою молекули 80 000 Да. Місце синтезу невідомо. У лабораторних умовах активується при зіткненні з поверхнею кварцу й скла, каоліну, целіту, азбесту, вуглекислого барію; а в організмі - при контакті зі шкірою, волокнами колагену, хондроїтинсірчаною кислотою, міцелами насичених жирних кислот, бактеріальними ліпополісахаридами, що містять радикали жирних кислот, ендотоксинами, адреналіном і норадреналіном. “Ініціатор” внутрішньосудинної коагуляції, активує прекалікреїни плазми, служить активатором фібринолізу. відіграє роль передавального механізму від системи згортання до калікреїн-кінінової системи, системи комплементу та фібринолізу. |
XIII, Фібринстабілізувальний фактор (трансглутаміназа)
|
α2-Глікопротеїд з масою молекули 300 000 — 340 000 Да. Виявляють в судинній стінці, тромбоцитах, еритроцитах, нирках, легенях, м'язах, плаценті. У плазмі перебуває у вигляді проферменту, з'єднаного з фібриногеном. Під впливом тромбіну перетворюється на активну форму (XIIIa). 10% фібринази забезпечують повноцінний гемостаз, 2% цього фактора достатні для зупинки кровотечі. Тромби, що утворилися в присутності фібринази, дуже повільно піддаються лізису. |
Фактор Фітцжеральда (кініноген)
|
Бере участь в активації фактора XI. У процесі згортання крові повністю не використовується. Його активність не знижується під впливом непрямих антикоагулянтів.При його відсутності порушується активація внутрішньої системи каоліном. |
Фактор Флетчера (прекалікреїн)
|
Бере участь у реакціях коагуляції в контактній фазі. При його відсутності порушується загальний час згортання. Активує фактори VII й IX, зв'язуючи внутрішню й зовнішню системи активації фактора X. |
Більшість факторів згортання є протеолітичними ферментами, що синтезуються у формі неактивних попередників - зимогенів. Вони активуються шляхом обмеженого протеолізу. Частина факторів не має ферментативної активності, а діє як алостеричні активатори.
До вітамін К-залежних білків належать фактори II, VII, IX, X, а також білки С i S. Вони утворюються в печінці, їхньою характерною ознакою є наявність карбоксиглутамінової кислоти (глу). Залишки глу, які утворюються внаслідок посттрансляційного карбоксилювання глутамінової кислоти за участі вітаміну К, групуються в особливих білкових доменах.
Приєднання додаткових, негативно заряджених карбоксильних груп потрібне для зв’язування факторами цієї групи іонів Са2+. Саме таке зв’язування забезпечує взаємодію між молекулами в реакціях плазмокоагуляції, які відбуваються за наявності іонів Са2+
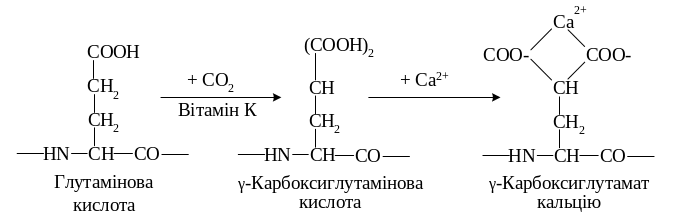
У разі нестачі вітаміну К в організмі утворюються дефектні молекули факторів II, VII, IX, X без глутамінової кислоти, які дуже повільно перетворюються на активні форми. Унаслідок цього сповільнюється згортання крові, спостерігається підвищена кровоточивість.
До патологій, пов’язаних з дефектами утворення вітамін К-залежних факторів згортання крові, належать гемофілія В (хвороба Крістмаса) та гемофілія Лейден пов’язані зі спадковим дефіцитом фактора IX, хвороба Стюарта-Прауера зумовлена недостатністю фактора X, спадкова гіпоконвертинемія - дефіцитом фактора VII, гіпо-(дис-) протромбінемія - зниженням синтезу або аномалією фактора II, а також комбінованим спадковим дефектом вітамін К-залежних факторів. Ці захворювання проявляються кровоточивістю мікроциркуляторного типу (петехії, синці), носовими кровотечами та кровотечами з ясен, тяжкими кровотечами після хірургічних втручань тощо.
Препарати групи кумарину (неодикумарин, фепромарон, нітрофарин, синкумарол тощо) інгібують вітамін К-залежне карбоксилювання залишків глутамінової кислоти в N-кінцевій частині молекули факторів II, VII, IX і X. Препарати кумарину застосовують для тривалого зниження згортання крові й лікування тромбозів, тромбофлебітів, тромбоемболічних ускладнень при інфаркті міокарда, емболічних уражень різних органів. Після введення вітаміну К синтез у печінці глу-вмісних факторів згортання відновлюється.
1
Рис.
12.10. Основні етапи коагуляційного
гемостазу
фаза активації – багатоступінчастий етап згортання, що завершується активацією протромбіну (фактор II) з перетворенням його на активний фермент тромбін (фактор IIа) (рис. 12.10);
фаза коагуляції – кінцевий етап згортання, у результаті якого під впливом тромбіну фібриноген (фактор I) перетворюється на фібрин.
Фаза активації. Найскладнішим і найдовшим процесом у цій фазі є формування протромбінази. Вона являє собою ферментний комплекс, що складається з активованих факторів згортання Ха, Va, іонів Са2+ і фосфоліпопротеїнів (фосфо-ЛП), джерелом останніх можуть бути:
фосфо-ЛП, що вивільняються при ушкодженні тканин, зокрема, ендотелію судин або сполучної тканини (тканинний тромбопластин - фактор III).
фосфо-ЛП мембран тромбоцитів, що виходять у плазму при їх руйнуванні (тромбоцитарний фактор 3).
Таким чином, формування ключового ферментного комплексу цієї фази відбувається двома шляхами, відповідно до яких розрізняють
зовнішню систему, яка активується при ушкодженні тканин впродовж 5 - 10 секунд: фосфо-ЛП, що виходять із тканинних клітин (тканинний тромбопластин, або фактор III), у присутності іонів Са2+ активують фактор VII (проконвертин) (рис. 12.11). Останній у комплексі з фосфо-ЛП ушкодженої тканини й іонами Са2+, у свою чергу, активує фактор Х, що входить потім до складу “протромбінази”.
внутрішню систему, активація якої відбувається трохи повільніше (впродовж 5 – 7 хвилин) і без участі тканинного тромбопластину. Пусковим фактором цього механізму є фактор XII (фактор Хагемана), який активується або при контакті крові з колагеном субендотелію ушкодженої судини чи з будь-якою чужорідною поверхнею (склом, металом тощо), або при ферментативному розщепленні фактора Хагемана протеолітичними ферментами (калікреїном (КК), тромбіном, трипсином тощо) за участі високомолекулярного кініногену (ВМК).
Фактор Хагемана (фактор XII) є універсальним активатором всіх плазменних протеолітичних систем - згортальної, калікреїн-кінінової, фібринолітичної і системи комплементу.
Фактор ХIIа активує фактор XI. Останній, у свою чергу, активує фактор IX. Нарешті, фактор IХа утворює ферментний комплекс з фосфо-ЛП, який у присутності іонів Са2+ і плазменного фактора VIIIа активує фактор X. останній - це Са2+-залежний глікопротеїн, який складається з легких і важких поліпептидних ланцюгів, які з'єднані дисульфідними місточками. Він синтезується в печінці за участі вітаміну К і являє собою серинову протеїназу. Активуючи фактор V, фактор Ха разом з ним входить до складу протромбінази.
Утворена двома шляхами протромбіназа протеолітично розщеплює неактивний попередник протромбін (фактор II) (молекулярна маса 72 000), у результаті чого утворюється активний протеолітичний фермент тромбін (молекулярна маса 35 000), що являє собою пептидазу. Дія тромбіну не обмежується лише протеолізом фібриногену на наступному етапі згортання крові, тромбін сприяє необоротній агрегації тромбоцитів, а також активує низку факторів згортання (V, VIII, XIII) (див. рис. 12.11, пунктирні стрілки).

Рис. 12.11. Взаємозв’язок між зовнішнім і внутрішнім шляхами згортання крові
фібриноген – глікопротеїн з молекулярною масою близько 340 000 Да, що складається з 2 946 послідовних амінокислот, і являє собою димер, у кожній одиниці якого містяться три поліпептидні ланцюги, з'єднані дисульфідними містками. Склад поліпептидних ланцюгів молекули фібриногену позначають Аα2, Вβ2 і γ2. Великі букви відповідають тим ділянкам, які від’єднуються від фібриногену під впливом тромбіну. Фрагменти А і В містять велику кількість аспартату та глутамату, що створює потужний негативний заряд на N-кінцях молекули фібриногену і запобігає агрегації (рис. 12.12). Фактор I у тому вигляді, у якому він виробляється паренхіматозними клітками печінки й надходить у кров, називається фібриногеном А, на відміну від фібриногену В, що осаджується із плазми вітаміном К (похідним β-нафтохінону).
Т
Рис.
12.12. будова
фібриногену: А і В – негативно заряджені
фрагменти, що запобігають агрегації;
D
і Е – глобулярні домени молекули
фібриногену, відділені ділянками
поліпептидних ланцюгів стрижнеподібної
конфігурації. З центрального домена
Е виступають N-кінцеві
ділянки фрагментів А і В ланцюгів Аα2
і Вβ2
Далі у фібринових згустках за участі фактора ХІІІа (трансглутамінази) та фібринази тканин, тромбоцитів, еритроцитів і Са2+ утворюються ковалентні зв'язки і фібрин S перетворюється на нерозчинний фібрин І (від англ. insoluble – нерозчинний). Внаслідок цього формується відносно м'який клубок ниток фібрину, в який потрапляють тромбоцити, еритроцити та лейкоцити, що призводить до їх руйнування.
Після утворення згустку починається процес ретракції, тобто ущільнення й закріплення тромбу в ушкодженій судині. Цей процес відбувається за допомогою скоротливого білка тромбоцитів тромбостеніну, іонів кальцію та енергії АТФ. Через 2-3 год згусток стискається до 25—50 % свого початкового об’єму. Завдяки ретракції тромб стає щільнішим і стягує краї рани. Ретрація триває від 30 хв до 30 год.
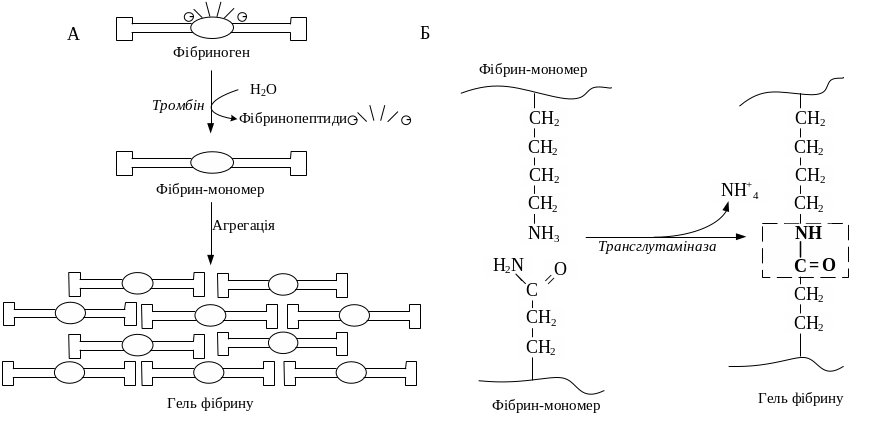 Рис.
12.13. Схема перетворення фібриногену
на фібрин
(А) та
виникнення амідних зв’язків між
молекулами фібрину (Б)
Рис.
12.13. Схема перетворення фібриногену
на фібрин
(А) та
виникнення амідних зв’язків між
молекулами фібрину (Б)
Варто згадати ще про один можливий шлях перетворення фібриногену на фібрин на кінцевій стадії згортання крові – про так званий «феномен паракоагуляції», що спостерігається, наприклад, при синдромі дисемінованого внутрішньосудинного згортання крові (ДВЗ-синдромі). На відміну від звичайного (описаного вище) процесу полімеризації волокон фібрину з його мономерів при цьому синдромі значно знижується чутливість до тромбіну й порушується процес полімеризації фібринів-мономерів. Це відбувається в результаті того, що частина фібринів-мономерів утворюють із фібриногеном і продуктами його розпаду комплексні крупно- і середньомолекулярні сполуки – розчинні фібрин-мономерні комплекси (РФМК). Вони погано реагують на дію тромбіну, володіючи відносною тромбінрезистентністю. Це і є феномен неферментативного згортання, або феномен паракоагуляції. Виявлення РФМК має важливе значення для діагностики ДВЗ-синдрому.
У результаті збільшення концентрації фібриногену в крові різко підвищуються ШОЕ, в'язкість крові, але не підсилюється гемокоагуляція. Зменшенням кількості фібриногену А нижче 1 г/л іноді обумовлені кровотечі внаслідок недостатності фібриногену (гіпофібриногенемия). Придбані гіпо- і афібриногенемії особливо часто виявляють акушери й хірурги у хворих на гострий ДВЗ-синдромом, рідше виникнення фібриногенопенії, пов'язані з тяжким порушенням функції печінки.
12.7.2.3. Порушення коагуляційного гемостазу. Гемофілія є спадковим захворюванням, зумовленим дефіцитом одного з про коагулянтів, які беруть участь в активації згортання крові. Виділяють гемофілію А (дефіцит фактора VІІІ), гемофілію В (дефіцит фактора ІХ) та гемофілію С (дефіцит фактора ХІ).
Найчастішою формою є гемофілія А, яка з загального числа хворих на гемофілію становить 87 – 94 % випадків. Успадковується гемофілія А по рецесивному, зчепленому з Х-хромосомою типу, у зв’язку з чим хворіють тільки чоловіки, а жінки є носіями гемофілії.
Гемофілію виявляють, зазвичай, у дитячому віці. Одним з характерних її проявів є крововиливи у великі суглоби кінцівок, утворення масивних підшкірних, внутрішньом’язових гематом, тривалі рецидивуючі кровотечі після травм та операцій, а також після незначних порізів шкіри та слизових оболонок, екстракції зубів та інших дрібних операцій.
Профузні шлунково-кишкові кровотечі виникають у хворих на гемофілію з супутньою виразковою хворобою, або після вживання ацетилсаліцилової кислоти. Перинатальна діагностика гемофілії А базується на аналізі ДНК у культурі клітин навколоплідних вод. Основним принципом лікування кровоточивості при гемофілії є введення в організм хворого достатньої кількості дефіцитного фактора.
А-(гіпо-)фібриногенемії характеризуються повною або частковою відсутністю в плазмі фібриногену. Патологія успадковується аутосомно-рецесивно, характеризується тяжкими кровотечами внаслідок повної відсутності здатності крові до коагуляції.
Дисфібриногенемії - коагулопатії, які виникають при амінокислотних замінах у первинній структурі молекул фібриногену. Аномальні молекули фібриногену мають змінену конформацію, що утруднює нормальний процес перетворення фібриногену на фібрин.
Тромбофілії – спадкові або набуті розлади гемостазу, наслідком яких є підвищена схильність до рецидивуючих тромбозів. Спадкові тромбофілії зумовлені спадковим дефіцитом антитромбіну ІІІ (АТ ІІІ), який інактивує тромбін, фактор Ха і, меншою мірою, фактори ХІІа, ХІа, ІХа. Швидкість інактивації значно зростає під впливом гепарину. Основним клінічним проявом даної патології є підвищена схильність до тромбозів різної локалізації. Перебіг захворювання залежить від ступеня дефіциту АТ ІІІ. Для легких форм дефіциту АТ ІІІ є характерним тромбування вен після звичайних внутрішньовенних ін’єкцій. При глибокому дефіциті АТ ІІІ тромбози можуть виникати внаслідок фізичного перевантаження, травми, вагітності, пологів тощо. Набуті тромбофілії зустрічаються частіше, ніж спадкові та зумовлені змінами ендотелію судин, активацією тромбоцитів, активацією згортання крові, гальмуванням дії фізіологічних інгібіторів згортання або системи фібринолізу. Спостерігаються при захворюваннях печінки, гемодіалізі, ДВЗ-синдромі, операціях на відкритому серці, при хіміотерапії.
12.7.3. Антизгортальна система крові. Крім речовин, які сприяють згортанню крові, існує низка інгібіторів згортання - антикоагулянтів, що запобігають гемокоагуляції.
За умов норми рідкий стан крові підтримується кількома механізмами (гладка поверхня ендотелію судин; негативний заряд стінки судин та формених елементів крові, внаслідок чого вони взаємовідштовхуються; наявність на стінці судин тонкого шару фібрину, який активно адсорбує фактори згортання, особливо тромбін; синтез ендотелієм простацикліну (потужного інгібітора агрегації тромбоцитів)), серед яких - постійна присутність у крові антикоагулянтів.
Серед фізіологічних антикоагулянтів виділяють: первинні - природно існують в організмі та вторинні - виникають в процесі згортання крові або під час фібринолізу. При деяких патологічних станах та під дією ліків з’являються патологічні антикоагулянти - імунні інгібітори окремих факторів згортання.
До первинних антикоагулянтів належать антитромбопластини, антитромбіни, гепарин. Антитромбопластини мають антитромбопластинову й антипротромбіназну активність. Антитромбіни зв’язують тромбін, з них найкраще вивчено антитромбін III.
За своєю природою антитромбін III (АТ-ІІІ) — глікопротеїн, який належить до α2-глобулінової фракції білків плазми крові. Його молекула складається з 432 амінокислотних залишків, має три дисульфідних зв’язки і 4 ділянки глікозилювання. Синтезується переважно клітинами паренхіми печінки, а також, незначною мірою, ендотелієм. У разі взаємодії АТ-ІІІ з тромбіном утворюється комплекс, в якому фермент й інгібітор сполучені ковалентним зв’язком ефірної природи. Формування комплексу відбувається зв’язуванням серину активного центру тромбіну із залишками аргініну молекули АТ-ІІІ, що супроводжується обмеженим протеолізом останнього. Він інгібує майже всі ферментні фактори згортання ІІа, Ха, ІХа, ХІа, ХІІа. АТ-ІІІ забезпечує 75 % всієї антикоагулянтної активності плазми крові, є основним плазмовим кофактором гепарину та слабким інгібітором плазміну і калікреїну. Природжене або набуте зниження рівня АТ-ІІІ спричинює розвиток тромбофілії з рецидивуючими тромбозами магістральних вен кінцівок та внутрішніх органів.
АТ-ІІІ зв’язує тромбін у співвідношенні 1:1, але активність антитромбіну ІІІ залежить від наявності гепарину, він має незначну ендогенну активність і не запобігає утворенню фібрину на місці кровотечі. Проте за певних умов у кровоплині з’являється активатор, який значно посилює активність АТ-III. Таким активатором є гепарин, що взаємодіє зі специфічною катіонною ділянкою АТ-III, спричинюючи конформаційні зміни його молекули.
У результаті таких змін АТ-III може зв’язуватися з сериновими протеїназами (переважно трипсином, а також хімотрипсином і плазміном). За фізіологічних умов гепарин зв’язується з АТ-III з високим ступенем спорідненості, їх взаємодія має електростатичну природу і залежить від рН середовища. У процесі утворення комплексу тромбін-антитромбін III останній виконує роль матриці для їх зближення, зв’язуючи молекулу ферменту з вільною ділянкою гепарину поруч з інгібітором, при цьому сам гепарин відіграє лише каталітичну роль. Після завершення активації тромбіну в потрійному комплексі АТ-III-тромбін-гепарин весь гепарин вивільнюється і зберігає свою біологічну активність. Таким чином, одна молекула гепарину може послідовно пришвидшити взаємодію багатьох пар молекул антитромбіну III і тромбіну.
При недостатності гепарину в крові активність АТ-ІІІ знижується в 30 разів, а при вмісті АТ-ІІІ менше 50 % від норми – гепарин повністю втрачає свої антикоагулянтні властивості. Це необхідно враховувати при проведенні гепаринотерапії.
Гепарин - природний сульфатований полісахарид, інгібітор полівалентної дії, (вперше його було виділено з клітин печінки, що і обумовило назву). Велика кількість гепарину інгібує всі фази згортання крові. Гепарин є не лише антикоагулянтом, він активує неферментний фібриноліз та поліпшує коронарний кровоплин.
За умов норми гепарин у крові не міститься. Він синтезується тканинними базофілами й базофільними лейкоцитами, особливо багато його в тканинах печінки, нирок, серця та м’язів. За деякими даними, на поверхні ендотелію судин міститься певна кількість глікозаміногліканів і глікопротеїнів, які мають гепариноподібні структури в складі своїх вуглеводних ланцюгів. Тому під час контакту з неушкодженим ендотелієм тромбін може інактивуватися антитробіном IIIза каталітичної дії глікопротеїнів із поверхні ендотелію.
Гепарин і фармацевтичні препарати на його основі (гепатромбін, гепароїд-мазь) застосовують для профілактики й терапії різних тромбоемболічних захворювань, тромбозів, тромбофлебітів тощо. Протизгортальну дію гепарину (наприклад, у разі його передозування) можна припинити катіонними пептидами. Такі поліпетиди конкурують із катіонними ділянками антитромбіну III за зв’язування з поліаніонним гепарином. Прикладом фармпрепаратів-антагоністів гепарину є протамін.
Протеїн С синтезується гепатоцитами, вітамін К-залежний профермент, який активується тромбіном. Після активації він розщеплює та інактивує основні неферментні фактори згортання крові (V і VІІІ). Вроджений дефіцит протеїну С спричиняє виникнення рецидивуючих тромбозів у дитячому та підлітковому віці.
α2-Макроглобулін – глікопротеїн α2–глобулінової фракції плазми крові, який є інгібітором протеїназ із широкою субстратною специфічністю, блокуючи серинові, карбокси- та металопротеїнази. Концентрація α2-макроглобуліну в плазмі крові людини найвища (до 2,5 г/л), порівняно з іншими протеїназними інгібіторами. α2–Макроглобулін є інгібітором тромбіну, його активність, на відміну від АТ ІІІ, не залежить від дії гепарину. На долю цього інгібітора припадає до 25 % антитромбінової активності плазми крові.
α1-Антитрипсин – глікопротеїн α1–глобулінової фракції плазми крові. Інгібітор має широкий спектр антипротеїназної дії, гальмуючи активність багатьох серинових протеїназ, зокрема тромбіну, факторів Ха і ХІа.
Прикладом вторинних антикоагулянтів може бути антитромбін І або фібрин, який адсорбує й інактивує тромбін. Продукти розпаду фібрину порушують полімеризацію фібрин-мономера, блокують фібрин-полімер і пригнічують агрегацію тромбоцитів. Сам тромбін також володіє антикоагулянтними властивостями. Діючи ферментативно на протромбін, він відщеплює від нього інгібітор фактора Ха.
Продукти ферментативного розщеплення фібрину, які утворюються під час фібринолізу, виявляють теж антикоагулянтні властивості, інгібуючи агрегацію тромбоцитів та утворення фібрину.
Патологічні антикоагулянти з’являються (часто в значних кількостях) при імунних порушеннях. До них відносять антитіла до факторів згортання крові VІІІ та V (характерно для гемофіліі, масивних гемотрансфузій і вживання деяких ліків).
Для процесу згортання крові характерне також явище самогальмування, тобто одні і ті ж самі фактори можуть спочатку виконувати коагулянтну функцію, а потім антикоагулянтну. Зокрема, фактор Vа після участі у згортанні починає гальмувати перетворення протромбіну на тромбін, а фактор ХІа - після взаємодії з фактором XII, IX гальмує ХІІа.
для консервування крові в якості антикоагулянта широко застосовують препарати солей лимонної кислоти (натрію цитрат), антизгортальна дія якого полягає в перетворені кальцію крові на цитрат кальцію. Це призводить до зв‘язування вільних іонів Са2+, які беруть участь в утворенні тромбопластину й перетворенні протромбіну на тромбін.
12.7.4. Фібринолітична система крові. Фібриноліз — процес ферментативного розщеплення фібрину кров'яного згустка, що супроводжується руйнуванням тромбу. За рахунок функціонування фібринолітичної системи відбувається постійне розчинення внутрішньосудинних тромбів, що можуть утворюватися на стінках кровоносних судин внаслідок дії факторів, які активують згортальну систему крові.
Активація фібринолізу в організмі людини, як і активація згортання крові відбувається за внутрішнім та зовнішнім механізмами.
Внутрішній механізм активації запускається тим самим фактором, що ініціює згортання крові, фактором ХІІа. Він взаємодіє з високомолекулярним кініногеном плазми та прекалікреїном, після чого набуває здатності активувати плазміноген. Може спостерігатися при цьому механізмі і самоактивація, внаслідок чого різко збільшується активність протеолізу фібрину.
Зовнішній механізм фібринолізу здійснюється білковими активаторами плазміногену, які синтезуються в судинній стінці (тканинні активатори). Інтенсивний викид активаторів у кров відбувається при порушенні прохідності судин, при фізичному навантаженні (стискання манжеткою), під впливом вазоактивних речовин та медикаментів (адреналіну, норадреналіну, нікотинової кислоти, аналогів вазопресину тощо). Потужні активатори плазміногену містяться у всіх клітинах крові, особливо в лейкоцитах. Крім цього, лейкоцити здатні продукувати внутрішньоклітинні кінази, які розщеплюють фібрин без участі плазміну. Містяться активатори плазміногену і в різних тканинах та секретах (слині, жовчі, молоці, сечі тощо). Велика кількість активаторів продукується клітинами деяких пухлин (меланома).
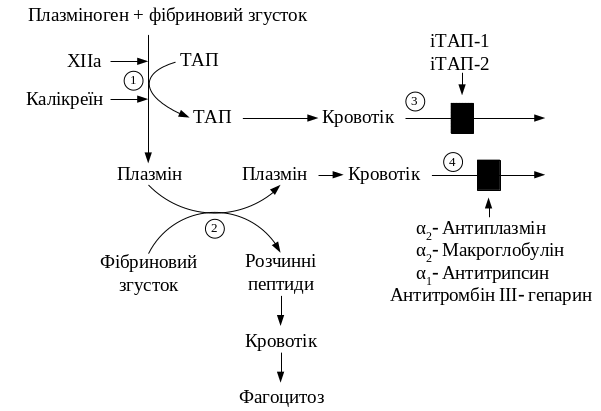
Фібриноліз складається з двох послідовних етапів:
І
Рис.
12.14. Схематичне зображення процесу
фібринолізу: 1- абсорбований на
фібриновому згустку плазміноген під
впливом активаторів перетворюється
на плазмін шляхом обмеженого протеолізу;
2 – плазмін гідролізує фібрин з
утворенням розчинних пептидів; 3 – у
крові ТАП інактивується специфічними
білками (іТАП-1, іТАП-2); 4 – активність
плазміну знижується під впливом
неспецифічних інгібіторів серинових
протеїназ
Плазміноген — глікопротеїн із класу β-глобулінів (М.м. приблизно 80 кДа), складається з одного поліпептидного ланцюга. Активація плазміногену з утворенням активного плазміну здійснюється за рахунок розщеплення протеїназами внутрішнього пептидного зв'язку Arg560 → Val561. Фізіологічними активаторами цього глікопротеїну є фактор ХIIа, судинні та тканинні активатори плазміногену (ТАП). Важливим активатором плазміногену є урокіназа, яка синтезується в нирках. Здатність активувати глікопротеїн без розщеплення внутрішньомолекулярного пептидного зв'язку має стрептокіназа — білок, що входить до складу β-гемолітичного стрептокока.
Плазмін — фермент, що за механізмом ферментативної дії є сериновою протеїназою трипсиноподібної дії, він може існувати в двох формах:
високомолекулярній - має кінцеву глутамінову кислоту (глу-плазміноген);
низькомолекулярній – внаслідок часткового лізису має кінцеву амінокислоту –лізин (ліз-плазміноген), активується швидше в 10 - 20 разів за глу-плазміноген.
II етап — розщеплення фібрину, що є основою фібринового згустка, до пептидних продуктів протеолізу. Процес каталізується активним плазміном, який утворився на І етапі фібринолізу. Адсорбування плазміногену на поверхні фібринових волокон відбувається ще під час утворення тромба. У молекулі плазміну є ділянки, комплементарні доменам фібрину, причому одна молекула плазміну може зв’язувати кілька молекул фібрину. Молекули ТАП також мають центри зв’язування з фібрином. Утворений з плазміногену плазмін під впливом ТАП гідролізує фібрин з утворенням пептидів X і Y, які активують фібриноліз, та D іE, які його гальмують. Ці пептиди потрапляють у кровотік і там фагоцитуються. Руйнування тромба призводить до вивільнення з нього плазміну та ТАП, які в крові легко ін активуються специфічними інгібіторами та захоплюються печінкою. Гальмування дії ТАП відбувається з допомогою і ТАП 1-ого і 2-ого типів, а плазміну - а2-антиплазміном.
а2-антиплазмін – одноланцюговий глікопротеїн з масою молекули 67 000, утворює комплекси з плазміном і трипсином. Взаємодія плазміну й α2-антиплазміну протікає в 2 стадії: стадію швидкого зворотного утворення фермент-інгібіторного комплексу й стадію його повільної трансформації в необоротний комплекс. α2-Антиплазмін взаємодіє також з фібрином. У результаті утворення комплексу фібринові згустки стають менш чутливими до фібринолізу плазміном. Спадкове зниження концентрації а2-антиплазміну в крові супроводжується підвищеною кровоточивістю.
До інгібіторів, які володіють слабшою дією, відносять α2 – макроглобулін, антитрипсин, С1- естеразний інгібітор тощо. При взаємодії з α2 – макроглобуліном плазмін уникає дії α2 – антиплазміну, але не може і впливати на фібрин.
У медичній практиці з метою лізису тромбів та профілактики тромбозів застосовують такі фармацевтичні препарати компонентів фібринолітичної системи: фібринолізин (плазмін), урокіназу, ТАП - серинова протеїназа, каталітично неактивна за відсутності контакту з фібрином. Перебуваючи в контакті з фібрином, ТАП може розщеплювати молекулу плазміногену з утворенням плазміну. Коли плазмін гідролізує фібрин, активатор плазміногену втрачає активність і протеоліз припиняється. Таким чином, забезпечується ефективна регуляція процесу фібринолізу. З метою терапії при інфаркті міокарда використовують ТАП, який сприяє відновленню прохідності коронарних артерій, зменшуючи, таким чином, ушкодження міокарда.
12.7.5. Роль пародонту в процесах згортання крові. Важливу роль у фізіології та патології пародонта відіграють процеси фібриноутворення та фібринолізу, оскільки в тканинах і рідинах порожнини рота присутні фактори гемокоагуляції та компоненти фібринолітичної системи, які визначають резистентність слизової оболонки рота.
Динамічні процеси фібриноутворення та фібринолізу на поверхні слизової оболонки порожнини рота певною мірою визначають гемостаз.
Активація цих процесів при запальних захворюваннях пародонту носить захисний характер. Відомо, що утворення фібрину відбувається при контакті тканин із рідким середовищем. яким є ендотеліальні клітини судин, поверхня серозних оболонок та епітеліальних покривів слизової оболонки порожнини рота. При запальних і деструктивних процесах у пародонті за рахунок зовнішнього механізму коагуляції швидко утворюється фібрин, тоді як після видалення зуба швидке розчинення згустка фібрину може сприяти кровотечі та протидіяти загоєнню, що може мати негативне значення для пацієнтів із геморагічними діатезами та іншими захворюваннями системи крові.
Джерелом фібринолітичної активності в тканинах ясен є ендотелій кровоносних судин пародонту, а також лізосоми епітеліальних клітин слизової оболонки порожнини рота і ясен. При запаленні пародонта вміст фібринолітично активних сполук у тканинах ясен зростає.