

Лекция № 1
Общая характеристика растворов. Межмолекулярные взаимодействия: ван-дер-ваальсовские и специфические. Уравнение Леннард-Джонса
Растворы, как физико-химические системы, широко распространены в природе и повсеместно используются в практике. Химически чистое вещество – это, по сути дела, некое идеальное предельное состояние, которое реально и практически не достижимо. Даже сверхчистые металлы, полученные в научных целях или в качестве стандартов и эталонов методом вакуумной и зонной плавки, содержат до 10-6% примесей, а значит являются твердыми растворами. Твердые, жидкие и газовые растворы играют исключительную роль в промышленности, в производстве жизненно важных химикатов, в катализе, в производстве самых современных материалов. Известные в настоящее время биологические процессы также протекают в растворах, а именно, в самом распространенном из них - воде. Все биологические жидкости (кровь, моча, пот, лимфа), в которых протекают биохимические процессы, таким образом являются водными растворами.
Раствор – это термодинамически устойчивая (равновесная) гомогенная система переменного состава, состоящая из двух или более компонентов.
Классификация: Растворы подразделяются на газовые, жидкие и твердые. Такая классификация конечно условная, однако она довольно полезна). Это представлено на слайде.
Газообразные (газовые) растворы – смесь газов, реже растворы твердых и жидких веществ в газах. В принципе, в определенных условиях газовые растворы способны распадаться на несколько фаз, например, смесь азота и аммиака – т.е., как это и не удивительно и противоречит здравому смыслу, газы могут быть ограниченно взаиморастворимы. Например, при 273К и 5000 атм система аммиак-азот распадалась на две газовые фазы. Одна фаза содержала 84.5% аммиака и 15.5% азота, а другая – 18% аммиака и 82% азота. В настоящее время известны многие системы, обладающие таким свойством: гелий - аммиак, диоксид серы - азот и т.д.
Важнейшии природные газовые растворы – воздух и природный газ. В состав воздуха входят азот (его больше всего), кислород (от него зависит жизнь на Земле), инертные газы (гелий, например, которые крайне важны в промышленности: сверхпроводимость при температуре жидкого гелия, инертная атмосфера – гелий, аргон). Для вас, как будущих нефтянников, важен такой газовый раствор, как природный газ, который, в основном, содержит метан). Составы этих газовых растворов приведены на слайде.
Химический состав воздуха в % на уровне моря при температуре 15 °c и давлении 101325 Па.
Газ |
Обозначение |
Процентное содержание |
Азот |
N2 |
78.084 % |
Кислород |
O2 |
20.9476 % |
Аргон |
Ar |
0.934 % |
Углекислый газ |
CO2 |
0.0314 % |
Неон |
Ne |
0.001818 % |
Метан |
CH4 |
0.0002 % |
Гелий |
He |
0.000524 % |
Криптон |
Kr |
0.000114 % |
Водород |
H2 |
0.00005 % |
Ксенон |
Xe |
0.0000087 % |
Источник: CRC Handbook of Chemistry and Physics by David R. Lide, Editor-in-Chief 1997 Edition
Состав природного газа (впервые установлен Клодом Бертолле)
Метан - 98,63%;
Этан - 0,12%;
Пропан - 0,02%;
Н-бутан - 0,1%;
Диоксид углерода - 1,01%;
Азот - 0,12%.
Жидкие растворы (в частности, водные) – наиболее часто встречающийся в практике тип растворов – это растворы в жидком растворителе твердых, газообразных и жидких веществ (или их комбинации). Различают растворы неэлектролитов и электролитов, высокомолекулярных соединений, органические и неорганические растворы. Хотя с термодинамической точки зрения все компоненты раствора равноценны, различают вещество растворитель (который после окончания процесса растворения остается в том же агрегатном состоянии) и растворенное вещество. Такое деление крайне условно (в зависимости от соотношения спирта и воды может быть раствор спирта в воде и наоборот), особенно если растворитель и растворенное вещество близки по природе и имеются в растворе в практически равных количествах, но тем не менее, такое деление полезно. Однако, в случае растворов электролитов в воде растворителем считается вода, независимо от концентрации (96% серная кислота).
Твердые растворы – это, например, сплавы металлов, керамики, ферриты, стекла. Т.Р. делятся на растворы замещения (1), внедрения (2) и вычитания (3).
Образование растворов замещения возможно, если оба компонента близки по кристаллохимическим свойствам и размеры частиц, занимающих узлы кристаллической решетки, отличаются не более чем на 15% (правило Юм-Розери), тогда атомы, ионы или молекулы одного вещества замещают в узлах кристаллической решетки частицы другого в-ва. При этом не меняется тип решетки и число атомов (ионов) в кристаллической ячейке (Z), но изменяются ее объем (V) и плотность (). Идентичность кристаллических решеток растворителя и растворенного вещества определяет возможность образования твердых растворов замещения любого состава. Примером такого твердого раствора является сплав меди и никеля. Атомы никеля способны занимают любые места в кристаллической решетке меди.
Если размеры частиц растворителя больше размеров частиц растворенного вещества, последние внедряются между узлами кристаллической решетки растворителя с образованием растворов внедрения (2). Такие растворы образуются при растворении неметаллов (бор, углерод, водород) в металлах. Наиболее распространенные растворы внедрения это раствор углерода в железе, а именно, разные композиции чугуна и нелегированной стали. При этом Z и V увеличиваются с ростом концентрации растворенного в-ва. При этом наблюдается увеличение напряжения в кристаллической решетке, поэтому область существования твердых растворов внедрения невелика. Концентрация растворов внедрения, обычно, тоже невелика.
Растворы вычитания (или твердые растворы с дефектной решеткой) (3) образуются за счет вакансий в кристаллической решетке, встречаются редко.
Различают твердые растворы с неограниченной и ограниченной растворимостью компонентов в твердом состоянии.
Термодинамические условия образования растворов.
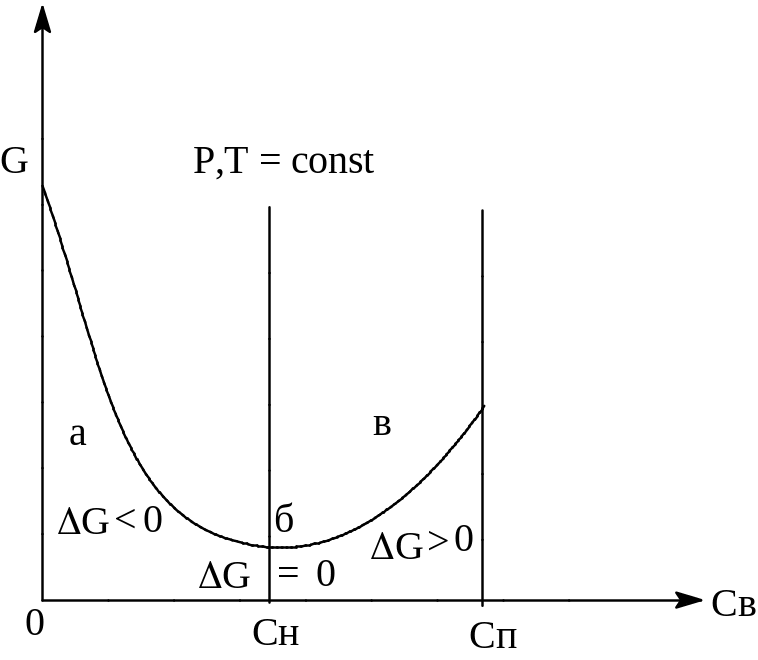
Каковы же термодинамические условия образования растворов. При внесении кристаллов в растворитель под действием молекул растворителя молекулы растворяемого вещества переходят в раствор. Затем вследствие диффузии (или перемешивания) частицы равномерно распределяются по всему объему растворителя. Раствор менее упорядоченная система, чем растворитель и растворенное вещество в отдельности. Это приводит к значительному росту энтропии при растворении твердых тел и жидкостей (при растворении газов – наоборот, энтропия уменьшается из-за того, что больший объем переходит в меньший объем) при растворении и убыли энергии Гиббса.
G H - TS (1)
Кроме того, процесс растворения обычно сопровождается значительным выделением тепла (экзотермический – вспомним растворение концентрированной серной кислоты в воде), т.е. H 0 (прирост энтальпии в изобарно-изотермических условиях равен количеству теплоты перешедшей к системе, если теплота уходит из системы, то изменение энтальпии меньше нуля), что еще более уменьшает энергию Гиббса при растворении.
Таким образом, растворение типично самопроизвольный (не требующий подвода энергии извне) процесс (знак равенства действует в случае равновесных процессов). Реже процесс растворения является эндотермичным (растворение нитрата аммония в воде), но и в этом случае энтропийный фактор превышает энтальпийный и все равно G 0. Таким образом, термодинамические соображения позволяют прогнозировать самопроизвольное растворение различных веществ на основе 1-го – закон сохранения энергии - U (изменение внутренней энергии) = Q (теплота)-A (работа) и 2-го начал термодинамики (изменение функции состояния S (энтропии) больше (неравновесный процесс) или равно (равновесный процесс) dS ≥ dQ/T).
 Повторю,
что чтобы судить о возможности
самопроизвольного протекания процесса
нужно учитывать два фактора: энергетический
и статистический. Система стремится к
минимуму потенциальной энергии и
максимуму беспорядка. Именно в связи с
самопроизвольностью процесса, чистые
вещества практически не встречаются в
природе.
Повторю,
что чтобы судить о возможности
самопроизвольного протекания процесса
нужно учитывать два фактора: энергетический
и статистический. Система стремится к
минимуму потенциальной энергии и
максимуму беспорядка. Именно в связи с
самопроизвольностью процесса, чистые
вещества практически не встречаются в
природе.
Можно записать дифференциальную форму предыдущего уравнения (1), где убыль энергии Гиббса G (эта величина комбинировано и сбалансировано учитывает и стремление системы к минимуму энергии, которая в условиях постоянства давления определяется энтальпией и стремление системы к максимальному беспорядку, выражаемому энтропией) выражена через химический потенциал компонента в кристалле (чистом виде) и растворе при растворении dni молей вещества:
dG = (р)dn - (к)dn 0 (2)
(к) (р) (3)
В равновесном состоянии при прекращении растворения:
(к) = (р) (4)
Влияние на растворимость природы компонентов (силы Ван дер Ваальса и «подобное в подобном»)
Хорошо известно, что вода растворяет спирт в любом количестве, тогда как с ртутью и нефтью она вообще не смешивается. Точно так же бензол растворяет углеводороды, но не растворяет воду. В чем причина этого феномена? Здесь можно дать такой общий ответ: жидкости смешиваются, если сходны их электронные структуры, а различия в электронной структуре затрудняют смешение. Чтобы пояснить, что мы понимаем под «электронной структурой», вновь рассмотрим воду. При образовании молекулы воды происходит перераспределение заряда между составляющими ее атомами: атомы водорода отдают свои валентные электроны, а атом кислорода принимает их. Таким образом, молекула воды имеет ненулевой электрический дипольный момент, т.е. является полярной. Этим объясняется, в частности, то, что вода обладает очень большой диэлектрической проницаемостью и соли хорошо растворяются в ней, диссоциируя на ионы. Диполь-дипольное взаимодействие удерживает молекулы воды вместе, вследствие чего повышается ее температура кипения. Другой пример полярной жидкости – спирт C2H5OH; он легко смешивается с водой, поскольку дипольный момент его молекул сходен с дипольным моментом молекул воды. Наряду с полярными жидкостями, молекулы которых в значительной степени связаны между собой, существуют и неполярные с более слабыми межмолекулярными связями. Примером таких жидкостей могут служить углеводороды – бензол, нафталин и др. Молекулы этих жидкостей построены из атомов углерода и водорода, которые обобществляют свои валентные электроны вместо того, чтобы отдавать или присоединять их. Об относительной слабости связей между молекулами углеводородов свидетельствует низкая температура их кипения. Между жидкостями с четко выраженными полярными свойствами (вода) и абсолютно неполярными (углеводороды) находится целый спектр классов жидкостей, так что не всегда можно заранее сказать, будут две данные жидкости смешиваться или нет. Но в большинстве случаев выполняется правило, сформулированное в начале раздела.
Кроме электронной структуры, смешиваемость жидкостей может существенным образом зависеть от размера молекул, а также от температуры. Например, никотин смешивается с водой в любой пропорции ниже 60° С и выше 208° С; при промежуточных же температурах взаимная растворимость никотина и воды весьма ограничена.
Обобщая, можно сказать, что способность веществ растворяться определяется характером межмолекулярных сил взаимодействия молекул компонентов раствора Х1 – Х2 в сравнении с их взаимодействиями в виде чистых веществ Х1 – Х1 и Х2 – Х2. Если эта сила для разнородных молекул увеличена по сравнению с соответствующими силами взаимодействия в чистых компонентах, то растворение происходит. Из практики известно, что наибольшая взаимная растворимость достигается, когда эти силы имеют сходную природу, т.е. справедливо старинное правило, известное еще алхимикам «подобное в подобном». Например, малополярные жидкости бензол и эфир неограниченно растворимы друг в друге. Соответственно, полярные вода и спирт также неограниченно растворимы друг в друге. Вещества с ионным типом связи хорошо растворяются в полярных растворителях (вода, спирты).
Исторически теория растворов начиналась с физической теории растворов (Вант-Гофф, Аррениус, Оствальд), которая рассматривала растворитель как инертную среду и приравнивала растворы к простым механическим смесям. В какой-то момент эта теория была основной и очень популярной, т.к. хорошо объясняла экспериментальные результаты, полученные при изучения коллективных (или коллигативных) свойств разбавленных растворов (осмос, эбулиоскопия, криоскопия, понижения давления пара над раствором – мы будем об этих явлениях говорить на последующих лекциях), которые не зависели от свойств растворенного вещества, а зависели только от природы растворителя и концентрации растворенного вещества. Т.е. считалось, что молекулы растворенного вещества в некотором приближении не взаимодействуют с молекулами растворителя и ведут себя подобно молекулам идеального газа. Так, (Якуб-Хендрикус) Вант-Гофф (голландский химик, долгое время работавший в Германии, Нобелевская премия 1901 г.) еще в октябре 1885 г. показал, что законы, найденные для давления газов, применимы и в случае очень разбавленных растворов, поведение которых близко к поведению идеальных растворов. Им был понят физиэческий смысл осмотического давления. Растворение рассматривалось как чисто физический процесс диффузии молекул растворяемого вещества в пустоты между молекулами растворителя. Повторю, что эта теория довольно хорошо описывала экспериментальные результаты. Даже аномальное поведение растворов электролитов, вследствие их распада их ионно или ковалентно построенных молекул на катионы и анионы в растворе, т.е. когда реальная концентрация частиц в растворе увеличена, было объяснено с помощью теории электролитической диссоциации (за это шведский физико-химик Сванте Аррениус в 1903 году получил Нобелевскую премию по химии – до него полагали, что диссоциация молекул на ионы происходит лишь под действием электрического тока). Распад молекул электролитов на ионы в растворителях с высокой диэлектрической проницаемостью (вода, формамид) называется электролитической диссоциацией. Небольшой экскурс в теорию электролитической диссоциацию.
В этом случае поправки в классические законы идеальных растворов вводили с использованием изотонических коэффициентов, которые учитывали распад молекул электролитов на ионы и реальное увеличение концентрации частиц в растворе. Однако, эта теория не могла объяснить ряда явлений, например, тепловые эффекты при растворении, свидетельствующие о взаимодействии молекул растворителя и растворенного вещества, т.к. тепловой эффект образования идеального раствора должен быть равен нулю H = 0, (дополнительные условия для идеального раствора V = 0, т.е. не наблюдается изменения объема и энтропия образования идеального раствора равна энтропии смешения компонентов идеального газа S = Sид.г. – Определение идеального раствора:
Идеальными будут такие растворы, образование которых из компонентов, взятых в одинаковом агрегатном состоянии и в любых соотношениях, не сопровождается изменением объема и тепловым эффектом, а изменение энтропии равно изменению энтропии при смешении идеальных газов.
Даже саму причину электролитической диссоциации физическая теория растворов, не учитывающая взаимодействия молекул, объяснить не могла.
Согласно химической теории растворов принципиально отличающейся от физической, при растворении вещества происходит его химическое взаимодействие с растворителем, с образованием неустойчивых динамических соединений, которые ассоциируют, диссоциируют, образуют молекулярные кластеры (сольваты, в частности, в водных растворах - гидраты), обменивающиеся с окружающей средой молекулами растворенного вещества и растворителя. В растворах вокруг молекул и ионов образуется оболочка, так называемая сольватная "шуба" из молекул растворителя. Химическая теория началась с Д.И. Менделеева, который на примере водных растворов показал, что при растворении сульфата меди, серной кислоты, спирта (зависимость первой производной плотности раствора по числу молей спирта в зависимости от состава раствора – эта кривая имеет ряд особых точек в виде переломов кривой, свидетельствующих об образовании молекулярных соединений спирта и воды состава С2Н5ОН 12Н2О, С2Н5ОН 3Н2О, 3С2Н5ОН Н2О – гидраты) и других веществ они образуют с водой соединения, называемые гидратами (сама теория поначалу тоже получила название гидратной теория). Менделеев резко критиковал чисто "физический" подход (в том числе и теорию Аррениуса) к пониманию природы растворов, не учитывающий химических взаимодействий между растворенным веществом и растворителем. Впоследствии выяснилось, что и Аррениус, и Вант-Гофф и Оствальд и Менделеев были каждый по-своему правы, и их взгляды, дополняли друг друга.
Межмолекулярные взаимодействия: ван-дер-ваальсовские и специфические.
Для объяснения поведения реальных растворов необходимо учитывать межмолекулярные взаимодействия, которые еще называют силами Ван-дер Ваальса.
Силы Ван-дер-Ваальса(СВДВ). Этот тип взаимодействия предложен голландским физико-химиком Ван-дер Ваальсом для объяснения поведения реальных газов и присущ всем молекулам. Изменение агрегатного состояния является проявлением этих сил. В частности, реальные газы при достаточно низких температурах и высоких давлениях сжижаются, что является проявлением СВДВ. Эти силы являются слабыми (их энергия 0,419–4,19 кДж/моль в сотни и тысячи раз меньше энергии химических связей, т.е. их действие не приводит к разрыву старых или образованию новых химических связей). Обобщенная теория сил Ван-дер-Ваальса достаточно сложна и, в общем случае, основывается как на электростатической теории, так и на квантовой механике. Молекулы веществ на малых расстояниях отталкиваются (силы отталкивания возникают в основном в результате перекрывания электронных оболочек), а на больших притягиваются.
Полная энергия межмолекулярного взаимодействия описывается формулой:
Евдв = Еор + Еинд + Едисп + Еотт
Ориентационное взаимодействие (электростатическое, эффект Кеезома). Определяется кулоновскими силами притяжения между дипольными молекулами. Схематически это взаимодействие можно представить картинкой, где полярные молекулы расположены линейно, (цепочечно, хвост в хвост). Ориентация дипольных молекул в кристаллах, на самом деле, может быть различной, (например, цепочечной или параллельной). Однако, эта ориентация является фиксированной. Для жидкостей и газов, для которых взаимная ориентация молекул в результате теплового движения непрерывно изменяется и в итоге усредняется, энергия взаимодействия между полярными молекулами A и B описывается формулой (знак «-» соответствует притяжения). Дипольные моменты имеют молекулы многих веществ: спирты, карбоновые кислоты, амины, нитросоединения. Этот тип взаимодействия уменьшается с увеличением температуры. В жидкой и газовой фазе ориентация нежесткая и нарушается (разрушается) тепловым движением с энергией kT (формула для ВВС в жидкости). 0 = 8.854188 10–12 Кл2/(Дж м) - диэлектрическая (электрическая) проницаемость вакуума.
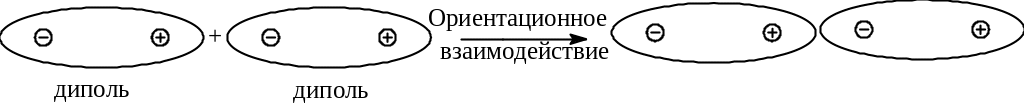
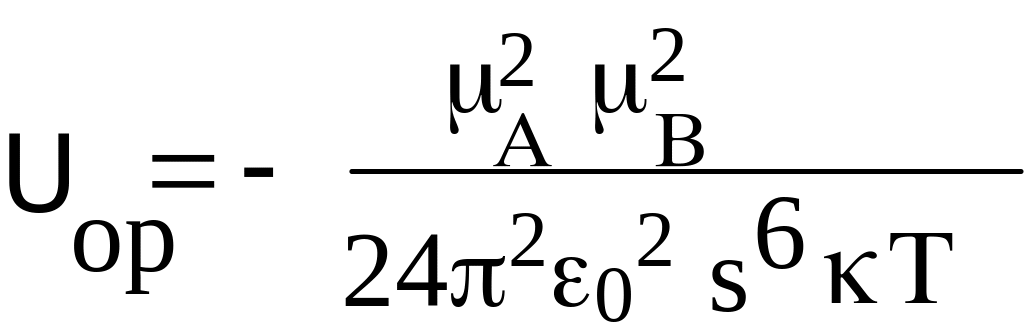
Индукционное
(поляризационное, эффект Дебая)
взаимодействие.
Возникает в результате разделения
(наведения) зарядов в неполярной молекуле
вследствие их поляризации (смещения
относительно друг друга, деформация
электронной оболочки поляризуемой
молекулы) зарядов под действием молекулы
с постоянным диполем. Это взаимодействие
не зависит от температуры, а зависит от
поляризуемости молекулы В и дипольного
момента м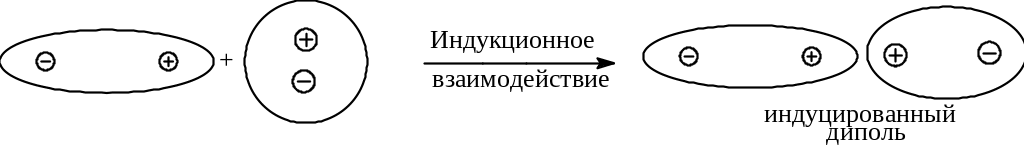
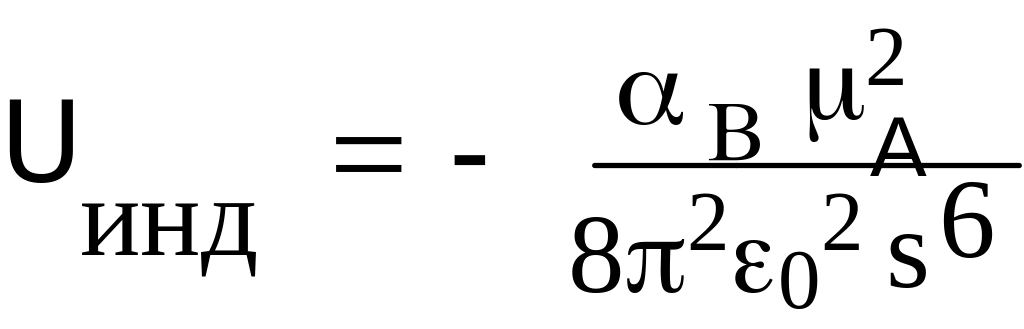 олекулы
А.
олекулы
А.
Дисперсионное (эффект Лондона) взаимодействие. Возникновение мгновенных диполей возможно между любыми молекулами, в том числе и неполярными. Причина этих сил – флуктуация распределения электронов в атомах и молекулах, в результате чего возникают мгновенные диполи. Так, молекулы с большим числом электронов и с более диффузным распределением электронов притягиваются сильнее, чем малые молекулы с более прочно связанными электронами. Например, в молекуле I2 106 электронов имеет более высокую температуру кипения (165оС), чем Cl2 (-35оС), в молекуле которого только 34 электрона. Другой пример, метан и бутан. Ясно, что молекулы бутана из-за диффузности распределения электронов в бутане сильнее притягиваются друг к другу и температура кипения бутана выше (-0.5оС по сравнению с метаном –164оС) Уравнение на основе квантово-механических представлений ввел английский физик Лондон. Зависит от потенциалов ионизации молекул A и B.
E дис
= -3/2 AB
(IA
IB/IA
+ IB)/s6
дис
= -3/2 AB
(IA
IB/IA
+ IB)/s6
I – потенциал ионизации, т.е. энергия, требующаяся для отрыва от молекулы одного электрона
Короткодействующие силы отталкивания определяются ядерным и электронным отталкиванием – они возрастают при сближении молекул очень быстро. Эти силы отталкивания являются причинами несжимаемости жидкостей.
П![]() отенциал
(уравнение) Леннард-Джонса
(Lennard-Jones
был первым профессором теоретической
физики в Великобритании. Он работал в
Университете Бристоля и покинул кафедру
в в 1932 г.). Интеллегентно запомнить, что
это был один человек, просто фамилия у
него двойная и по правилам русского
языка следует говорить «потенциал
Леннард-Джонса» - я даже в солидных
учебниках встречал «Леннарда - Джонса»
и даже «Леннарда-Джонсона».
отенциал
(уравнение) Леннард-Джонса
(Lennard-Jones
был первым профессором теоретической
физики в Великобритании. Он работал в
Университете Бристоля и покинул кафедру
в в 1932 г.). Интеллегентно запомнить, что
это был один человек, просто фамилия у
него двойная и по правилам русского
языка следует говорить «потенциал
Леннард-Джонса» - я даже в солидных
учебниках встречал «Леннарда - Джонса»
и даже «Леннарда-Джонсона».
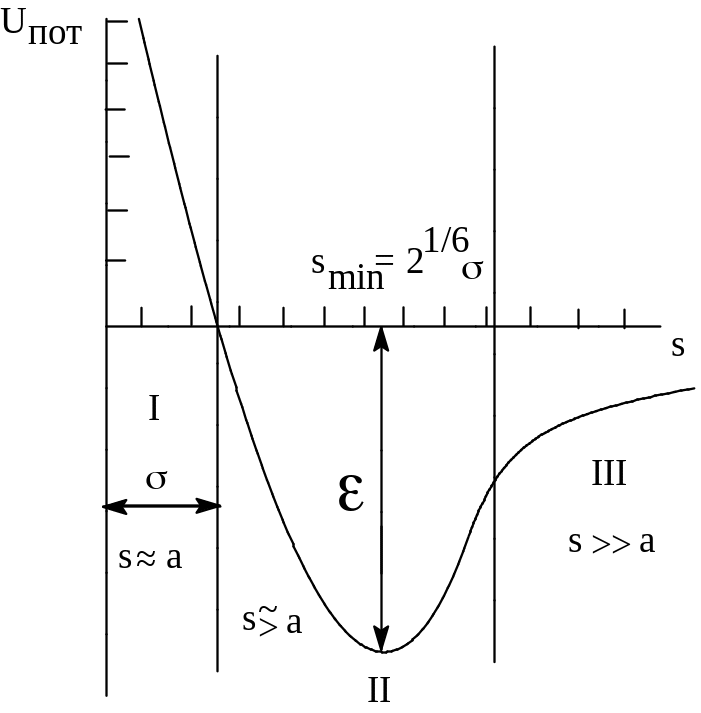 Потенциал
Леннард-Джонса может быть в обобщенном
виде записан формулой. Его графическое
выражение представлено на графике
(потенциал 6-12) (rmin
= :21/6,
= глубина минимума), где
- глубина потенциальной ямы или
ван-дер-ваальсова энергия взаимодействия
на равновесном расстоянии so
- эта величина достаточно мала 1-5 кДж/моль,
примерно в 100 раз меньше энергии химической
связи, равновесное расстояние so
3-5 ангстрем, значительно превышает
межядерное расстояние в молекулах и
(такое значение s
при котором Uпот=0)
являются специфическими параметрами,
определяемыми экспериментально
(например, через эксперименты с рассеянием
молекулярных пучков или явлений переноса
(диффузия, теплопроводность) в газах).
Их можно найти в таблицах. Например, для
воды
= 0.6501696 кДж/моль,
= 0.3165555 нанометров. Эти параметры находят
экспериментально при исследовании, в
основном, столкновений молекулярных
пучков, т.к. их рассеивание зависит от
межмолекулярного взаимодействия. Их
можно найти в таблицах. Область III, где
s
a (а - размер молекулы - все вещества
состоят из молекул - мельчайших частиц
материи, молекулы газов имеют средний
размер (радиус) ~ 10-8
см и массу ~ 10-22
кг – массу молекулы можно определить
поделив вес одного моля вещества на
число Авогадро NA=6*1023
моль-1
- число молекул в 1 моль вещества),
область
II, s несколько больше a, область I – где
s - сравнимо с а, и вступают в действие
короткодействующие силы отталкивания.
ВВС очень важны для понимания свойств
растворов, газов и для понимания
химических реакций на поверхности и в
катализе.
Потенциал
Леннард-Джонса может быть в обобщенном
виде записан формулой. Его графическое
выражение представлено на графике
(потенциал 6-12) (rmin
= :21/6,
= глубина минимума), где
- глубина потенциальной ямы или
ван-дер-ваальсова энергия взаимодействия
на равновесном расстоянии so
- эта величина достаточно мала 1-5 кДж/моль,
примерно в 100 раз меньше энергии химической
связи, равновесное расстояние so
3-5 ангстрем, значительно превышает
межядерное расстояние в молекулах и
(такое значение s
при котором Uпот=0)
являются специфическими параметрами,
определяемыми экспериментально
(например, через эксперименты с рассеянием
молекулярных пучков или явлений переноса
(диффузия, теплопроводность) в газах).
Их можно найти в таблицах. Например, для
воды
= 0.6501696 кДж/моль,
= 0.3165555 нанометров. Эти параметры находят
экспериментально при исследовании, в
основном, столкновений молекулярных
пучков, т.к. их рассеивание зависит от
межмолекулярного взаимодействия. Их
можно найти в таблицах. Область III, где
s
a (а - размер молекулы - все вещества
состоят из молекул - мельчайших частиц
материи, молекулы газов имеют средний
размер (радиус) ~ 10-8
см и массу ~ 10-22
кг – массу молекулы можно определить
поделив вес одного моля вещества на
число Авогадро NA=6*1023
моль-1
- число молекул в 1 моль вещества),
область
II, s несколько больше a, область I – где
s - сравнимо с а, и вступают в действие
короткодействующие силы отталкивания.
ВВС очень важны для понимания свойств
растворов, газов и для понимания
химических реакций на поверхности и в
катализе.
Энергия сил Ван-дер-Ваальса взаимодействия полярных и неполярных молекул
Моле-кула |
Поляри-зуемость, 1024 см3 |
Диполь-ный момент, 1028 эсе |
Энергия ионизации, эв |
Энергия
Ориента-ционная |
Взаимо-действия, Поляриза-ционная |
эрг 1012
дисперси-онная |
O2 |
1.74 |
0 |
15.8 |
0 |
0 |
57.20 |
Cl2 |
4.60 |
0 |
12.7 |
0 |
0 |
321.0 |
CO |
1.99 |
0.12 |
14.3 |
0.054 |
0.057 |
62.5 |
HBr |
3.58 |
0.78 |
13.3 |
6.2 |
4.05 |
176.0 |
HCl |
2.63 |
1.03 |
13.7 |
18.6 |
5.4 |
105.0 |
H2O |
1.48 |
1.84 |
18.0 |
19.0 |
10.0 |
47.0 |
Как это видно из таблицы, дисперсионные силы играют основную роль в межмолекулярном взаимодействии и являются единственными для неполярных молекул.
Строго говоря, этот потенциал не может абсолютно точно работать и описывать молекулярные ансамбли, т.к. учитывает взаимодействие только двух молекул. Уже третья молекула вблизи взаимодействующих двух будет изменять потенциал. Хотя такие подходы с использованием сложного математического аппарата и мощных вычислительных машин имеются, но учет многочастичного взаимодействия сильно усложняет теорию СВДВ и делает ее не такой наглядной. Поэтому, для наших целей мы будем пользоваться упрощенными подходами.
Ван-дер ваальсовы молекулы. Т.к. СВДВ невелики, то соединения за счет их действия при нормалных температурах не образуются. Этому препятствует тепловое движение. Но при очень низких температурах, когда энергия взаимодействие больше kT удается спектрально обнаружить соединения типа Ar2, Xe2, ArHCl, ArN2 и т.д.
Специфические формы межмолекулярного взаимодействия.
Водородная связь. (hydrogen bond) - слабая электростатическая связь, в результате которой атом водорода располагается между двумя электроотрицательными атомами (например, азота или кислорода). Большое количество водородных связей в белках и нуклеиновых кислотах приводит к наличию у этих соединений стабильной молекулярной структуры.
Молекула ДНК (дезоксирибонуклеиновая кислота) – носитель генетической (наследственной) информации – состоит из двух полинуклеотидных цепей, закрученных одна вокруг другой в спираль. Цепи удерживаются в виде спирали благодаря многочисленным водородным связям между составляющими их основаниями. Аналогично ведут себя и белки. В ряду соединений с водородом элементов VI группы – Н2О–Н2S–H2Se–Н2Те – температура плавления возрастает почти линейно для трех последних веществ. Если бы зависимость температуры плавления от номера периода сохранялась по всей группе, то лед плавился бы (пунктирная линия) примерно при температуре 170 К (–100 °С). Однако температура плавления льда нарушает ожидаемую закономерность – она почти на 100 °С выше (273 К, или 0 °С). По-видимому, это связано со значительно более прочной кристаллической решеткой льда по сравнению с кристаллическими решетками других гидридов.
Повышенная
прочность кристаллической воды
обусловлена двумя главными причинами:
первая
– угол между связями в молекуле воды
благодаря sр3-гибридизации
внешних электронных оболочек атома
кислорода близок к тетраэдрическому
углу (109,5°), в то время как в молекулах
остальных гидридов VI группы углы близки
к 90°; вторая
– образование водородных связей между
молекулами воды. Возникновение такой
связи обусловлено тем, что электронное
облако атома водорода в молекуле воды
смещено к ядру атома кислорода, благодаря
чему имеется дополнительная возможность
для взаимодействия протона с еще одним
атомом кислорода другой молекулы воды.
Таким образом, атом водорода в структуре
воды образует одну прочную ковалентную
связь с ближайшим атомом кислорода и
одну более слабую, водородную, связь с
атомом кислорода соседней молекулы
воды. Атом кислорода в молекуле воды
имеет две sр3-гибридные
орбитали, заполненные парами электронов,
которые он и поставляет для образования
двух водородных связей. Таким образом,
каждая молекула воды связана с четырьмя
другими молекулами воды. Следовательно,
водородная связь между молекулами воды
обусловлена акцепторным характером
протона и донорным поведением отрицательно
заряженного атома кислорода. В
кристаллической структуре льда каждая
молекула воды образует четыре связи с
соседними молекулами воды за счет своих
двух атомов водорода и двух гибридных
орбиталей атома кислорода, имеющих по
паре электронов. Все четыре связи между
молекулами кристаллической воды
равноценны. Структуру льда можно
представить в виде структуры алмаза,
если атомы углерода заменить молекулами
воды. Угол между связями всех молекул
воды почти равен тетраэдрическому.
Водородные связи проявляются также в
жидкой воде. Вода кипит при сравнительно
высокой температуре (100 °С), что указывает
на высокий уровень порядка в жидкой
воде, вызванный тем, что в ней существуют
«осколки льда» (Н2О)n,
состоящие из сотен молекул воды. Даже
в газовой фазе ассоциация молекул воды
сохраняется, и там обнаружены частицы
из нескольких молекул в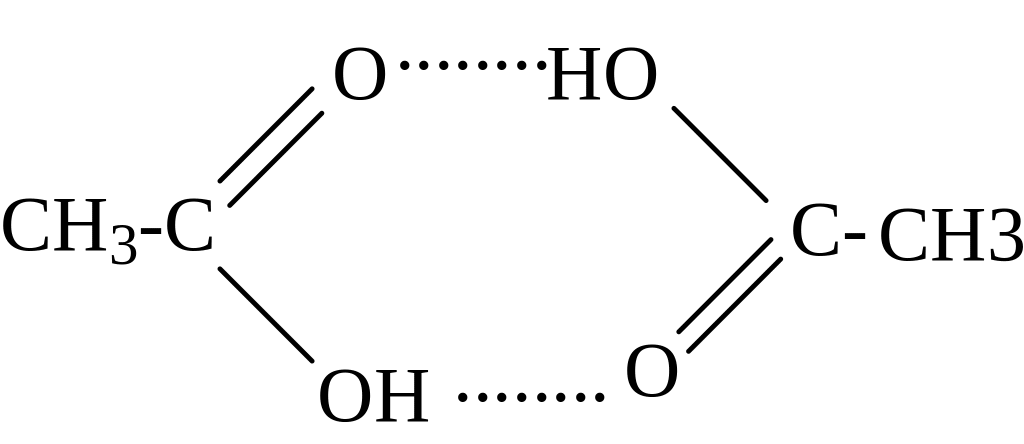 оды.
Такова прочность водородных связей
между молекулами воды! Водородные связи
о
оды.
Такова прочность водородных связей
между молекулами воды! Водородные связи
о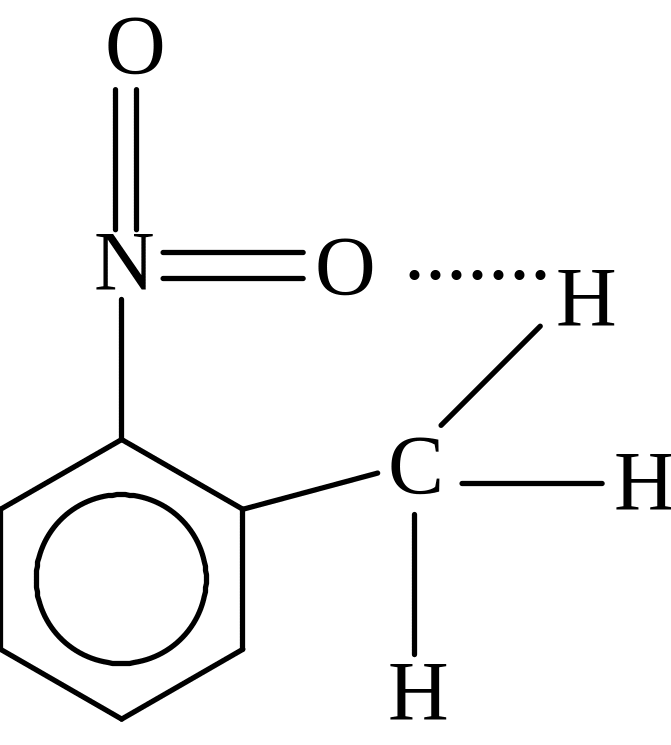 бразуются
также между молекулами аммиака NH3
и фтороводорода HF, но структуры этих
веществ (кристалл, жидкость) менее
прочны. Это вызвано не слабостью
водородных связей между их молекулами,
а их числом. В жидком аммиаке от каждой
молекулы исходит три связи, что
обусловливает плоскостную структуру
связанных между собой молекул. В структуре
жидкого фтороводорода одна молекула
HF связана с двумя другими, и молекулы
образуют зигзагообразные, вытянутые в
линию цепочки. Почему же молекулы Н2S,
H2Se,
Н2Те
и многие другие, как образующие, так и
вообще не образующие водородных связей,
могут существовать в жидком и
кристаллическом состояниях? Что связывает
эти молекулы между собой? В этих веществах
значительную роль играют силы
Ван-дер-Ваальса,
силы притяжения электростатической
природы между частицами. Аналогичную
воде аномалию имеет NH3
в пятой группе и HF
в седьмой.
бразуются
также между молекулами аммиака NH3
и фтороводорода HF, но структуры этих
веществ (кристалл, жидкость) менее
прочны. Это вызвано не слабостью
водородных связей между их молекулами,
а их числом. В жидком аммиаке от каждой
молекулы исходит три связи, что
обусловливает плоскостную структуру
связанных между собой молекул. В структуре
жидкого фтороводорода одна молекула
HF связана с двумя другими, и молекулы
образуют зигзагообразные, вытянутые в
линию цепочки. Почему же молекулы Н2S,
H2Se,
Н2Те
и многие другие, как образующие, так и
вообще не образующие водородных связей,
могут существовать в жидком и
кристаллическом состояниях? Что связывает
эти молекулы между собой? В этих веществах
значительную роль играют силы
Ван-дер-Ваальса,
силы притяжения электростатической
природы между частицами. Аналогичную
воде аномалию имеет NH3
в пятой группе и HF
в седьмой.
Карбоновые кислоты, спирты, фтористоводородная кислота – вот примеры, когда между молекулами возникает специфическое взаимодействие, называемое водородной связью, через водородный мостик, которая является более прочной, чем ВДВ взаимодействие, но менее прочной, чем химическая связь (энергия 20-40 кдж/моль). Энергия водородной связи составляет около 5 ккал/моль. Эта оценка базируется на сравнении экспериментальных теплот испарения сходных соединений, из которых одни могут завязывать водородные связи, а другие — нет). Например, для карбоновых кислот в растворах характерны димеры. Акцептором электронов является протонизированный водород (Н+), а донором электронов являются атомы, несущие несвязанную (неподеленную) электронную пару (ОН, NO2, SH и т.д.). Водородная связь может быть как меж-, так и внутримолекулярной (хелатной). Водородная связь играет исключительную роль в химии и биологии (структура белка и ДНК – нити двойной спирали связаны водородным связями).
Ниже приведены температуры кипения некоторых веществ.
Вещество |
формула |
t , °C |
Ацетон |
СН3СОСН3 |
58 |
Этиловый спирт |
С2Н5ОН |
78 |
Вода |
Н2О |
100 |
Олеиновая кислота |
С17Н33СООН |
~270 |
Глицерин |
СН2ОНСНОНСН2ОН |
290 |
Можете ли вы сказать, какие из перечисленных веществ в жидком состоянии имеют наиболее ММВ, включая сильные водородные связи?
Существуют и другие типы достаточно сильного взаимодействия в растворах:
Ионно-молекулярное взаимодействие
Сольваты в полярных растворителях: например, при растворении литиевыех (другие щелочные металлы) солей в полярных растворителях Li+ (ТГФ)n
Аквакомплексы: (Cr3+ (H2O)6), ион гидроксония Н3О+,
Гидроксокомплексы: (CeOH3+)
Комплексы с переносом зарядов: пара-ксилол с CBr4, Ag+ C6H6
Резюмируем то, что о чем мы с вами сегодня говорили. Таким образом, в соответствии с современными представлениями,
Растворение – сложный физико-химический процесс, в котором играют роль как физические (ион-дипольная при растворении веществ с ионной структурой, диполь-дипольные при растворении вещест с молекулярной структурой - органических), так и химические (донорно-акцепторные связи – аква-комплексы, водородные связи) взаимодействия.
Т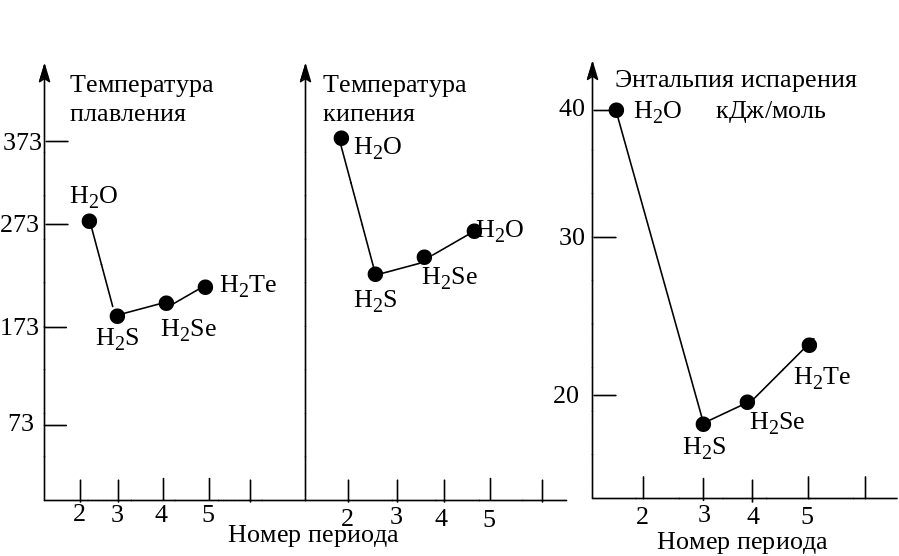 емпература
плавления и кипения, а также энтальпия
испарения гидридов элементов VI
группы
емпература
плавления и кипения, а также энтальпия
испарения гидридов элементов VI
группы