

l6. ОСМОТИЧЕСКОЕ ДАВЛЕНИЕ
В 1748 г. А. Нолле впервые наблюдал, как растворитель проходит через мембрану из разбавленного раствора в более концентрированный. Если к более концентрированному раствору приложить давление, то в зависимости от его величин течение растворителя может быть замедлено или остановлено. Это явление было названо осмосом.
Осмотическим давлением раствора называется то наименьшее давление, которое помимо давления самого растворителя необходимо приложить к раствору, чтобы предотвратить перемещение растворителя к раствору через мембрану, разделяющую раствор и растворитель, причем мембрана непроницаема для молекул растворенного вещества.
Иллюстрацией осмотического давления может служить рис. 69. В левом отделении прибора находится чистый растворитель, в правом — раствор. Эти две жидкости отделены друг от друга полупроницаемой мембраной (например, мембраной из целлофана). Поры в мембране достаточно велики, чтобы через них свободно проходили молекулы растворителя, но мембрана препятствует проникновению молекул растворенного вещества из правой части сосуда в левую. Скорость перехода растворителя из левой части сосуда в правую в начальный момент больше, чем скорость его перемещения в обратном направлении. Поэтому высота столба жидкости в правой части сосуда увеличивается до тех пор, пока не будет достигнуто равновесие, при котором разность уровней жидкости в левой и правой частях (h) отвечает осмотическому давлению.
В 1877 г. В. Пфеффер измерил осмотическое давление π нескольких растворов, приготовленных путем растворения одной и той же массы вещества в разных объемах растворителя. При этом он показал, что если поддерживать температуру постоянной, то произведение πV всегда будет одним и тем же. Для данного раствора с повышением температуры осмотическое давление увеличивается, причем отношение π/Т сохраняется постоянным. Я. Вант-Гофф обобщил эти результаты и предложил эмпирическое управление для описания осмотического давления растворов:
π = СRT,где π - осмтичесое давление; С- молярная концентрация раствора; Т-температура.
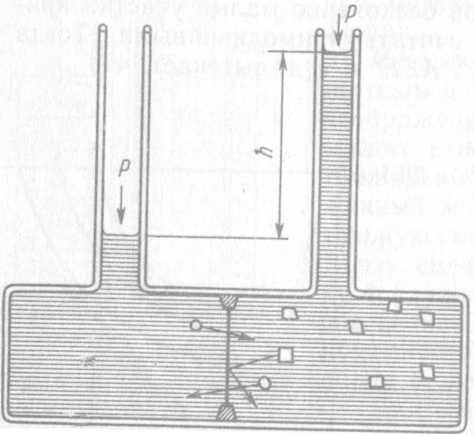
Рис. 69. Прибор для демонстрации осмотического давления.
кружки - молекулы растворителя; квадратики- молекулы растворённого вещества.
Линейная зависимость осмотического давления от концентрации раствора и от температуры соблюдается только для идеальных растворов. Поэтому уравнение (7.4) можно применять только для разбавленных растворов. Если растворенное вещество диссоциирует и имеет степень диссоциации а (см. гл. VIII, разд. 2.1), то в простейшем случае диссоциации одной частицы на две имеем
АВ↔А+ + В-
Число недиссоциированных частиц, получившихся из 1 моль, равно (1—а) моль, число продуктов диссоциации равно 2а моль, а всего молей будет
1 — а + 2а=1+a.
Сумму 1 +а обозначают буквой j. Это так называемый изотонический коэффициент Вант-Гоффа (см. гл. VIII).
Тогда уравнение осмотического давления (7.3) принимает вид
n = iCRT.
Осмотическое давление можно определять двумя основными методами: статическим и динамическим.
Статический метод основан на том, что осмотическое давление раствора уравновешивается давлением столба жидкости, возникающем в результате проникновения растворителя в раствор.
Осмометр (рис. 70) состоит из камеры 2 вместимостью ~ 10 мл из стекла или хромированной латуни. Камера присоединяется с помощью винтов к пластинке, в середине которой имеются отверстия (диаметром 1 мм). Нижняя сторона камеры плотно прижимает мембрану к пластинке — сетке, толщина которой должна быть не больше 0,5 мм. Часть осмометра, в которой находится раствор, называется осмотической ячейкой. Раствор наливается в ячейку через верхнее отверстие У, куда для отсчета давления вставляется пришлифованный градуированный капилляр диаметром 1 мм и длиной 50 см. Нижняя пришлифованная часть капилляра входит в камеру на 0,5... 1 мм для предохранения камеры от пузырьков воздуха при заполнении.
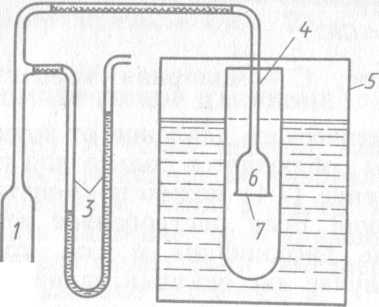
Рис. 71. Осмометр для определения осмотического давления динамическим методом
Камера с раствором вставляется в сосуд 3, наполненный чистым расворите-лем. Сосуд закрывается пришлифованной крышкой во избежание испарения растворителя. При измерении осмотического давления осмометр помещают в термостат.
Динамический метод основан на том, что осмотическое давление компенсируется наложенным на раствор переменным противодавлением. Осмотическое давление вычисляется на основании измерения скорости проникновения растворителя через мембрану. Преимущество динамического метода заключается в быстроте измерений.
На рис. 71 представлен компенсационный осмометр А. В. Думанского. Осмотическая ячейка 6 соединяется с мембраной 7, аспиратором 1 и манометром 3. Ячейку и внешний сосуд помещают в термостат 5. Поднятием верхней части аспиратора 2 регулируется внешнее давление таким образом, чтобы оно было больше или меньше осмотического давления. При избыточном внешнем давлении мениск в капилляре 4 опускается вниз со скоростью υ1, пропорциональной избыточному давлению р—π. При пониженном внешнем давлении мениск поднимается со скоростью υ2, пропорциональной разности π—ρ2 тогда отношение скоростей перемещения растворителя по капилляру υ1/υ2 будет равно отношению разностей давлений, обусловливающих поднятие или опускание мениска в капилляре, т. е.
υ1/υ2=ρ1-π/π-ρ2
отсюда
π=υ1ρ2+υ2ρ1/υ1+υ2
Осмотическое давление данного раствора может быть экспериментально определено по понижению температуры замерзания или по повышению температуры кипения раствора.
Предполагая равенство моляльной и молярной концентрации (m≈С) для разбавленных растворов, можно при сопоставлении уравнений
ΔТ=Кm, π= CRT
получить значение осмотического давления, связанного с величиной ДГ, которое определяют экспериментально:
Аналогично можно установить связь осмотического давления с повышением температуры кипения и эбуллиоскопической константой.
Осмотическое давление играет большую роль в жизни клеток. Каждая растительная клетка покрыта прочной целлюлозной оболочкой, к которой плотно прилегает протоплазма клетки. Поверхностный слой этой протоплазмы обладает свойствами полупроницаемой оболочки и, следовательно, свободно пропускает воду и не пропускает или почти не пропускает многие растворенные в воде вещества. Целлюлозная оболочка свойствами полупроницаемости не обладает и поэтому легко проницаема для всех растворенных веществ.
Если растительную клетку перенести в концентрированный раствор какого-нибудь вещества (например, сахара или хлорида натрия), молярная концентрация которого будет выше, чем концентрация растворенных веществ в клетке, то наблюдается осмотическое высасывание воды из клетки в окружающий ее внешний раствор (экзосмос). Протоплазма уменьшается в объеме и отстает от стенок целлюлозной оболочки. Объем протоплазмы делается тем меньше, чем большей концентрации был раствор, в который погружена клетка. При соответствующих условиях протоплазма принимает шарообразную форму, уменьшаясь в несколько раз. Это явление называется плазмолизом. Если плазмоли-зированную клетку поместить снова в раствор обычной для нее концентрации или в дистиллированную воду, клетка, благодаря осмотическому всасыванию растворителя, увеличивает свой объем, возвращаясь в свое исходное положение. Таким образом, плазмолиз является обратимым процессом.
Если нормальную растительную клетку поместить в раствор, концентрация которого будет ниже, чем концентрация растворенных веществ в самой клетке, или просто в дистиллированную воду, то под влиянием более высокого осмотического давления содержимого клетки происходит осмотическое всасывание воды в клетку (эндосмос). Объем клетки при этом увеличивается, растягивая стенки целлюлозной оболочки: клетка при этих условиях находится в состоянии напряжения. Состояние осмотического напряжения клетки, обусловленное повышенным осмотическим давлением, называется тур горком, Он поддерживает в напряжении ткани и органы у растений. Увядание растений связано с уменьшением тургора.
Естественно, что клетки окажутся неизменными, если их поместить в среду, осмотическое давление которой одинаково с внутриклеточным осмотическим давлением. Все упомянутые осмотические процессы присущи также животным тканям и клеткам.
Растворы, осмотическое давление которых одинаково с осмотическим давлением клеток и тканей, называются изоосмотическими или изотоническими. Растворы, молярная концентрация которых, а стало быть, и осмотическое давление, выше, чем внутри клеток и тканей, называются гипертоническими. Растворы, молярная концентрация которых, а следовательно, и осмотическое давление, ниже, чем в клетках и тканях, называются гипотоническим и..
Итак, если изотонические растворы не вызывают изменений в клетках, то растворы гипертонические обусловливают явление плазмолиза, а растворы гипотонические — явление тургора.
Нормальные растительные клетки всегда тургоризированы. Иначе говоря, концентрация растворенных веществ клетки выше, чем концентрация растворенных веществ в окружающей среде. Тургор растительных клеток является необходимым условием для роста и нормальной жизнедеятельности.
В растениях наблюдается значительное осмотическое давление, достигающее 0,5...2,0 МПа. Некоторые растения пустынь и засоленных почв, которым приходится особенно упорно бороться за влагу, имеют осмотическое давление, достигающее 5 МПа и даже 17 МПа.
Особенно высоко осмотическое давление в точках роста. Так, у клеток стеб
левых узлов злаков оно достигает 5 МПа. корни всегда имеют более высокое осмотическое давление, чем почвенный раствор, откуда они всасывают воду и питательные вещества.
2.1. Слабые электролиты. Степень диссоциации
Процесс диссоциации слабых электролитов является обратимым. Молекулы распадаются на ионы, а образующиеся ионы противоположного знака, встречаясь в растворе, могут вновь соединяться в молекулы. Для электролита вида АВ процесс диссоциации можно записать так:
А
 В
А+
+
В-
В
А+
+
В-
Как и во всяком обратимом процессе, здесь устанавливается некоторое динамическое равновесие. Так как слабые электролиты подчиняются, закону действующих масс, это равновесие может количественно характеризоваться константой равновесия, называемой в данном случае константой диссоциации. Для рассматриваемого электролита, распадающегося на два иона (так называемый бинарный электролит), эта константа выразится соотношением
[А+] [В-]
К= _________
[АВ]
Следует подчеркнуть, что эта константа может достаточно корректно характеризовать лишь разбавленные растворы слабых электролитов.
Количественная характеристика равновесного состояния диссоциации слабого электролита может быть получена также с использованием степени диссоциации а, которая показывает, какая часть молекул электролита в растворе распалась на ионы:
Число распавшихся молекул
а=_____________________________
Общее число растворенных молекул
Значение а всегда меньше единицы. Умножая а на 100, получают долю ионизированных молекул в процентах.
Степень диссоциации связана с константой диссоциации. Так, для бинарного электролита АВ концентрации ионов [А+] и [В+] связываются с общей концентрацией электролита С соотношением
[А+] = [B+] = аС.
Степень диссоциации зависит от природы электролита, природы растворителя, температуры раствора и степени его разбавления. Как уже было рассмотрено, чем больше диэлектрическая постоянная растворителя, тем значительнее при прочих равных условиях электролитическая диссоциация растворенного в нем вещества. Диссоциация молекул — процесс эндотермический. Следовательно, по принципу Ле Шателье с повышением температуры степень диссоциации должна возрастать. Однако при повышении температуры уменьшается диэлектрическая постоянная растворителя, что благоприятствует образованию молекул из ионов. У большинства электролитов степень диссоциации по мере повышения температуры увеличивается, а у некоторых (например, NH4OH, СН3СООН) достигает максимума, а затем уменьшается.
С разбавлением раствора электролита вероятность взаимодействия ионов в растворе уменьшается, степень электролитической диссоциации увеличивается. При этом константа диссоциации, вычисленная для данного слабого электролита, в сильно разбавленном растворе имеет постоянное значение (табл. 27).
Таблица 27. Значение а и К для уксусной кислоты при различных разбавлениях
Разбавление, м3/кмоль |
А·102 |
К·105 |
13,57 |
1,570 |
1,845 |
27,14 |
2,216 |
1,851 |
108,56 |
4,380 |
1,849 |
868,4 |
11,90 |
1,850 |
3474,0 |
22,36 |
1,855 |
Экспериментальные данные свидетельствуют, что растворы электролитов обнаруживают всегда большие отклонения от свойств чистого растворителя, чем растворы неэлектролитов. Отклонения определяются большим числом (концентрацией) частиц растворенного вещества в растворе электролита вследствие диссоциации его молекул на ионы. В результате диссоциации число частиц оказывается больше расчетного. Следовательно, в уравнениях, которые выражают зависимость свойств раствора от его концентрации для неэлектролитов, требуется ввести поправочный множитель, отражающий это явление
Оценка отклонения позволяет определять степень диссоциации. Поэтому значение степени диссоциации может быть определено по величине осмотического давления, понижению температуры замерзания раствора и данным электрической проводимости.
Если в растворе до диссоциации находилось N молекул и если степень диссоциации их при данных условиях равна а, то число диссоциированных молекул будет
N-Na = N( 1- а).
Когда каждая молекула способна распадаться на n ионов, число всех частиц (молекул и ионов) равно
N(1-a) + nNa = N(1-а +nа).
Осмотическое давление пропорционально числу частиц, следовательно, наблюдаемое осмотическое давление πопр пропорционально общему числу частиц (молекул и ионов) после диссоциации, т. е. числу N(1 — а + nа). Вычисляемое же осмотическое давление πвыч пропорционально числу частиц (молекул) до диссоциации, т. е. N. Отсюда
N(1— а -nа)
J=-------------------=1 +а(n-1) (8.5)
N
ИЛИ
j-1
а=------------
n-1
Уравнение (8.5) позволяет понять физический смысл изотонического коэффициента.
Изотонический коэффициент показывает, во сколько раз суммарная эффективная концентрация недиссоциированных молекул и ионов больше чем концентрация молекул до диссоциации.
В частном случае для бинарного электролита (n = 2)
i =1 + а, а = i — 1.
Таким образом, определив осмотическое давление πопр экспериментально и вычислив πвыч из формулы π = iCRT, можно получить степень диссоциации слабого электролита.
Другим методом экспериментального определения степени диссоциации является криоскопический метод, основанный на определении кажущейся молекулярной массы М1, которая представляет собой среднее значение масс ионов и недиссоциированных молекул:
i = М/М1,
где М — истинная молекулярная масса, откуда
М/М1-1
а=-----------------
n-1
а для бинарного электролита
М — M1
а =----------------
M1
5. ИОННОЕ ПРОИЗВЕДЕНИЕ ВОДЫ. ПОНЯТИЕ рН
При количественном определении кислотности водных растворов рассматриваются только разбавленные водные растворы, т. е. такие растворы, в которых количество вещества (молей) кислоты или основания, растворенных в литре воды, гораздо меньше количества вещества (молей) воды (приблизительно 55,5 кмоль/м3). Тогда можно считать, что количество вещества (молей) воды, например, в 1 л остается практически постоянным, несмотря на химические реакции, которые могут протекать в растворе, приводя к распаду или возникновению молекул воды. Рассмотрим диссоциацию самой воды. Равновесие диссоциации воды выражается уравнением
Н
 2О
+ Н2О
Н3О+
+ОН
2О
+ Н2О
Н3О+
+ОН
Диссоциация воды очень мала, поскольку при 298 К константа равновесия
[Н3О+] [ОН-]
К=---------------------------=3,24.10-18
[Н2О]
Концентрацию воды Н20 в разбавленном растворе можно считать постоянной. Тогда
1000
[Н20] == --------------- 55,5 кмоль/м3.
18
Концентрации ионов Н3О+ и ОН-при этом равны, так как раствор электронейтрален. Тогда,
[Н30+] [ОН-] = 55,52-3,24·10-18 (кмоль/м3)2 = 10-14 (кмоль/м3)2 = Kw,
[НзО+] = [ОН-] = 10 кмоль/м3,
где Kw — ионное произведение воды.
Так как Kw≠ 0, не может быть водного раствора, в котором концентрация ионов Н30+ или ОН-равнялась бы нулю. Следовательно, в любом водном растворе всегда присутствуют совместно ионы Н30+ и ОН-
Следует также отметить, что диссоциация воды является эндотермическим процессом, а ее образование из ионов — экзотермическим. Отсюда в соответствии с принципом Ле Шателье температура будет оказывать значительное влияние на Kw (табл. 29).
Таблица 29. Зависимость Kw и рН от температуры
т, к |
Kw |
РН |
||
273 |
1,139· |
10 |
-16 |
7,97 |
291 |
5,702· |
10 |
-15 |
7,11 |
298 |
1,008· |
10 |
-14 |
6,99 |
323 |
5,474· |
10 |
-14 |
6,63 |
373 |
5,90· |
10 |
-13 |
6,12 |
Раствор называется кислым, если концентрация ионов Н3О+ выше 10-7 кмоль/м3, а концентрация ионов ОН- ниже этого значения. И наоборот, щелочной раствор соответствует концентрации ионов ОН- выше 10-7 кмоль/м3 при концентрации ионов Н30+ ниже этого значения. Если концентрации ионов Н30+ и ОН- равны 10-7 кмоль/м3, то раствор называется нейтральным.
Разбавленные растворы кислот всегда соответствуют очень низким концентрациям ионов Н30+, обычно выраженным отрицательной степенью 10. Для краткого выражения концентрации Зёренсен ввел в 1920 г. новое понятие, известное под названием рН.
Величина рН раствора определяется как отрицательный десятичный логарифм числа, выражающего концентрацию ионов Н30+ в этом растворе.
Итак,
1
pH = -lg[H30+]=lg-------------
[Н3О+]
Но концентрации ионов Н30+ и ОН- водного раствора всегда должны удовлетворять равновесию диссоциации воды:
[Н30+] [ОН-] = Kw= 10-14 (кмоль/м3)2.
Отсюда вытекает следующая связь между концентрациями ионов Н30+ и ОН-водного раствора:
lg [Н30+] + lg [ОН-] =-14.
Тогда, воспользовавшись аналогичным понятием рОН = — lg [ОН -], можно записать
рН + рОН= 14.
На самом деле значением рОН не пользуются, а вычисляют рН щелочного раствора из соотношения
рН=14-рОН.
рН кислых и щелочных водных растворов рассчитывают по-разному, исходя из силы кислоты или основания по отношению к воде. Поэтому необходимо рассмотреть классификацию кислот и оснований по отношению к воде.
7. БУФЕРНЫЕ СИСТЕМЫ
Буферными называются растворы, представляющие собой смесь слабой кислоты и одной из ее солей с сильным основанием или слабого основания и одной из его солей с сильной кислотой.
В качестве смеси слабой кислоты и соли слабой кислоты и сильного основания рассмотрим раствор, концентрация уксусной кислоты которого равна С1 кмоль/м3, а концентрация ацетата натрия С2 кмоль/м3 .
Равновесие диссоциации уксусной кислоты в водном растворе
1
С
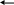 НзСООН
+ Н20
СНзСОО-
+ Н30+
(8.22)
НзСООН
+ Н20
СНзСОО-
+ Н30+
(8.22)
2
соответствует слабой диссоциации; оно смещено в направлении
2. В растворе имеется также ацетат натрия, который реагирует с водой:
1
С Н3СОО- +Н2О СНзСООН+ОН- (8.23)
2
Равновесие (8.22) характеризуется константой кислотности Ка:
[СНзСОО-] [Н30+]
Ка=---------------------------------------------
[СНзСООН]
Следовательно, концентрация ионов НзО+ в растворе равна
[СНзСООН]
[НзО+]=--------------------- (8.24)
[СНзСОО-]
Итак, для расчета рН необходимо знать концентрации уксусной кислоты и ацетат-ионов в растворе при равновесии.
Рассмотрим влияние друг на друга равновесных систем (8.22), (8.23). Введение в раствор ионов СН3СОО- в форме соли вызывает смещение равновесия (8.22) в направлении 2; в присутствии избытка ацетат-ионов ионизация уксусной кислоты становится еще слабее, так как добавление общего иона приводит к уменьшению степени диссоциации, Поэтому степень диссоциации а достаточно мала, что позволяет пренебречь концентрацией диссоциированной уксусной кислоты по сравнению с начальной концентрацией кислоты. Тогда можно записать
[СН3СООН] = С1(1 -а)≈С1.
Гидролиз ацетата приводит к возрастанию концентрации уксусной кислоты в данном растворе. Однако присутствие уксусной кислоты подавляет гидролиз, и равновесие (8.23) смещается в направлении 2. Поэтому количеством уксусной кислоты, образовавшейся при гидролизе, можно также пренебречь по сравнению с количеством кислоты, введенной первоначально в раствор. Отсюда следует, что
[СНзСООН]≈C1. (8.25)
Концентрация ацетата-ионов будет определяться следующими факторами:
1) ацетат натрия полностью диссоциирован в воде и концентрация ацетат-ионов, первоначально введенных в раствор в виде соли, равна С2;
2) уменьшение концентрации ацетат-ионов, вызванное гидролизом соли, достаточно мало при избытке кислоты и им можно пренебречь;
3) уксусная кислота при избытке ацетат-ионов диссоциирует довольно слабо, чтобы можно было пренебречь концентрацией ацетат-ионов, возникающих при диссоциации кислоты, по сравнению с начальной концентрацией ацетат-ионов С2. Тогда можно считать, что
[СНзСОО-]≈С2. (8.26)
и с учетом (8.25) и (8.26) выражение (8.24) для концентрации ионов Н30+ примет вид
С1
[Н30+] =Ка--------
С2
Таким образом, рН легко определить, зная начальные концентрации кислоты и соли в растворе:
С2
pH=pКа + lg--------
С1
т. е. в общем случае
[СОЛЬ]
pH-=pКa+lg---------
[кислота]
При рассмотрении смеси слабого основания и соли слабого основания и сильной кислоты ход рассуждений такой же, как и для раствора, содержащего слабое основание и одну из солей сильной кислоты. Если взять в качестве примера водный раствор аммиака и хлорида аммония, то реакция между основанием NH3 и водой представлена равновесием
N
 H3+Н20
NH4+
+ ОН-
H3+Н20
NH4+
+ ОН-
а реакция гидролиза соли — равновесием
N H4+ + Н20 NH3 +Н3О+
К онцентрация ионов Н30+ и рН раствора определяется из соотношения
[NH4+]
[НзО+]=Ка-------------
[NH3]
где Ка — константа кислотности пары NH4+/NH3.
Воспользовавшись для определения равновесных концентраций теми же приближениями, что и в предыдущем примере, можно получить
[ NH3] = С1
[NH4+]= С2,
где С1 и С2 — начальные концентрации NH3 и NH4C1 соответственно. Имеем
С2
[Н30+] = Ка--------
С1
Если концентрации основания и соли равны, то
[Н30+] = Ка, рН = рКа.
В общем случае
С1 [основание]
рН = рКа + lg--------,т.е.рН =рКа + lg-----------
С2 [соль]
Свойства буферных растворов. В противоположность растворам слабой кислоты и слабого основания рН этих растворов не
зависит от концентрации, если не учитывать коэффициенты активности компонентов.
Действительно, как уже было показано, для двух типов буферных растворов рН выражается соотношениями
[кислота]
[НзО+] = Ка ------------, [НзО+] = Ка-----[соль]-------
[соль] [основание]
При разбавлении раствора меняется концентрация кислоты и соли или основания и соли, но их отношения остаются постоянными.
Важнейшее свойство буферных растворов заключается в том, что их рН мало изменяется при добавлении умеренных количеств сильной, кислоты или сильного основания.
Рассмотрим буферный раствор, состоящий из смеси уксусной кислоты и ацетата натрия, и рассчитаем изменение рН, вызванное прибавлением 10-2 кмоль соляной кислоты к 1 м3 раствора, содержащего 1 кмоль уксусной кислоты и 1 кмоль ацетата натрия. Соляная кислота, полностью ионизированная на ионы Н30+ и С1-, реагирует с ацетатом натрия:
1
С Н3СООН- + Н3О СНзСООН + Н20
 2
2
Это равновесие сильно смещено в направлении 1; соответствующая константа равновесия равна
________[СНзСООН]___=__1_=5.5 •104
[СН3СОО-] [Н30+] Ка
Ка
Следовательно, практически образуется 10-2моль уксусной кислоты и исчезает 10-2моль ацетата. В этих условиях концентрация ионов Н30+ раствора равна
[кислота] 1+0,01
.[Н3О+]=Ка---------- = Ка--------
[соль] 1 — 0,01
т. е. изменение составляет только 2% кислоты и соли. Для рН получаем
1,01
рН = рКа- lg-------- = 4,75 - 0,13 = 4,62.
0,99
Это значение сравнимо со значением, соответствующим исходному раствору, содержащему 1 кмоль/м3 уксусной кислоты и ацетата натрия (рН = рКа = 4,75).
Таким образом, изменение рН незначительно. Необходимо отметить, что прибавление 10-2кмоль соляной кислоты в 1 м3 чистой воды изменит рН раствора с 7 до 2, т. е. на 5 порядков.
Прибавление сильного основания к буферному раствору также лишь слегка изменит количество образовавшейся соли и уменьшит количество кислоты, присутствующей в растворе, взяв только очень небольшое изменение рН. Можно показать, что сильнее всего буферное свойство проявляется для раствора эквимолекулярных количеств кислоты и соли или основания и соли.
Чем меньше изменение рН при добавлении в буферный раствор кислоты или основания, тем сильнее выражено буферное свойство раствора.
Интервал, в котором проявляется буферное свойство, называется буферной емкостью.
Она определяется количеством сильной кислоты или основания (в кмолях), которое необходимо добавить в 1 м3 буферного раствора, чтобы сместить рН на единицу:
СV
В=---------------
(рН1-рН2) Vбуф
где В — буферная емкость; С — концентрация сильной кислоты или основания, кмоль/м3; V — объем добавленной кислоты или основания, м3; Кбуф — объем буферного раствора, м3; pH1 и рН2 — водородные показатели соответственно до и после добавления сильной кислоты или основания.
Биологическое значение буферных растворов. Буферные системы почв.
Буферные растворы особенно важны для биологических сред, в которых рН должен оставаться неизменным. Так, например, рН крови должен быть постоянным и близким 7,4. Однако питание и обмен веществ могут вызвать приток кислых или щелочных соединений. Для поддержания рН крови постоянным в биологических средах существуют системы эффективных регуляторов, состоящие из буферных растворов, например смесей NaHCО3 — Na2CО3 или NaH2P04 — Na2HPO4.Наряду с этими химическими простыми буферными системами крови следует отметить так называемый гемоглобиновый буфер, играющий важную роль в поддержании постоянства рН крови, так как он обеспечивает около 75% буферной емкости крови. Гемоглобиновый буфер представляет собой смесь калиевой соли гемоглобина и свободного гемоглобина, являющегося слабой органической кислотой.
В живых клетках постоянство рН обеспечивается так называемым белковым буфером, который состоит из протеина и его натриевой соли.
Почва и почвенные растворы также обладают определенной буферной характеристикой, которая обусловливается наличием коллоидов и поглощенных катионов. Большое значение имеют энергия поглощения водородных ионов коллоидами и степень диссоциации коллоидов. Органическое вещество почвы преимущественно состоит из слабых кислот, поэтому оно увеличивает буферность почвы.
Почвенный раствор обладает буферным свойством по отношению к кислотам, если в нем присутствуют соли сильных оснований и слабой кислоты. К сильным основаниям относятся натрий, калий, к менее сильным — кальций и магний. Слабые кислоты в почве представлены гуминовыми кислотами, фульвокислотами, щавелевой, угольной и др. Сильные — серной, азотной, соляной. Последние частично попадают в почву с удобрениями либо освобождаются при поглощении растениями питательных веществ из физиологически кислых удобрений, например аммония из сульфата аммония.
Буферную характеристику почв необходимо учитывать в сельскохозяйственном производстве. Так, в слабобуферных почвах реакция может резко измениться от внесения кислых удобрений, что влияет на развитие растений. Хорошие буферные свойства по отношению к кислотам имеют почвы, богатые органическими коллоидами (черноземы, луговые, некоторые торфяные), минеральными коллоидами (суглинистые, глинистые), содержащие много поглощенных оснований. Почвы, бедные органическими и минеральными коллоидами (щебнистые, песчаные), содержащие много поглощенного водорода и мало кальция и магния, обладают плохой буферной характеристикой. Кислые почвы, например подзолистые, имеют хорошую буферную характеристику по отношению к щелочам и плохую по отношению к кислотам.
Улучшение буферной характеристики почв осуществляют внесением органических или минеральных коллоидов и ила; в кислых почвах это достигается внесением извести. Кислые минеральные удобрения в почвы, не обладающие буферными свойствами, вносят небольшими дозами или одновременно с известью. Особенно важна эта осторожность в начальный период развития растений.
АДСОРБЦИЯ. АДСОРБЦИЯ НА ГРАНИЦЕ ТВЕРДОЕ ТЕЛО — ГАЗ
Любая гетерогенная система, т. е. система, обладающая поверхностью раздела фаз, характеризуется определенным запасом поверхностной энергии. Эта энергия тем больше, чем больше поверхность раздела фаз. Так как разные поверхности обладают различной поверхностной энергией, то для их сравнения используют понятие удельной поверхностной энергии — энергии, приходящейся на единицу площади поверхности.
В изотермических условиях и при постоянном объеме изменение энергии поверхности равно изменению изохорно-изотерми-ческого потенциала:
F = óS,
где F — изохорно-изотермический потенциал, отвечающий общей энергии поверхности; S — площадь поверхности; ó — удельная (приходящаяся на 1 м2) поверхностная энергия
Согласно второму началу термодинамики процессы, идущие с уменьшением потенциала F, протекают самопроизвольно. В гетерогенных системах изохорно-изотермический потенциал может уменьшиться либо при сокращении поверхности S, либо при уменьшении поверхностной энергии о. Поверхность твердых тел самопроизвольно не уменьшается, она постоянна. Тогда самопроизвольное уменьшение поверхностной энергии может быть только результатом снижения поверхностной энергии, а при поглощении молекул газа или растворенного вещества поверхностью твердого тела.
Поглощение каким-либо веществом других веществ называется сорбцией.
Если процесс сорбции идет только на поверхности, то его называют адсорбцией, которая представляет собой увеличение концентрации вещества на границе раздела фаз. Если поглощаемое вещество диффундирует в глубь поглотителя и распределяется по объему, то это явление называется абсорбцией.
То вещество, на поверхности которого идет адсорбция, принято называть адсорбентом, а вещество, которое адсорбируется, — адсорбатом. Адсорбцию Г обычно выражают соотношении адсорбата х приходящимся на единицу площади поверхности адсорбента S, в кмоль/м2:
Г = х/S.
Если адсорбентом является твердое пористое тело, общую поверхность которого определить невозможно, то адсорбцию Г относят к единице массы адсорбента в кмоль/кг:
Г = х/m.
Адсорбция может идти на поверхности раздела следующих фаз: газ — твердое тело, раствор — твердое тело, газ — раствор.
Адсорбцию газа углем наблюдал К. Шееле еще в XVIII в. На явление же адсорбции веществ из раствора впервые обратил внимание русский акад. Т. Е. Ловиц (1785). Н. Соссюр (1814) определил, что все пористые тела, т. е. тела с большой удельной поверхностью, способны адсорбировать газы и что при этом обычно выделяется теплота. Отсюда следует, что процесс адсорбции является экзотермическим процессом.
Описание взаимодействия молекул адсорбата и молекул адсорбента представляет собой весьма сложную и до сих пор нерешенную задачу. Силы взаимодействия адсорбента и адсорбата, определяющие адсорбцию, различны, и обычно рассматривают два крайних случая, когда адсорбция характеризуется физическими либо химическими взаимодействиями: так называемая физическая или химическая адсорбция.
Ф и з и ч е с к а я адсорбция возникает за счет ван-дер-ваальсовых взаимодействий. Она характеризуется хорошей обратимостью, отсутствием стехиометрических соотношений, уменьшением адсорбции при повышении температуры, близостью эффектов адсорбции к теплотам снижения или испарения (обычно 10... 80 кДж/моль). Такова, например, адсорбция благородных газов на угле.
X и м и ч е с к а я адсорбция (хемосорбция) осуществляется только путем химического взаимодействия. Здесь адсорбция почти необратима, тепловой эффект близок к энергии образования химических соединений.
Так как хемосорбция является химическим процессом, требующим энергии активации порядка 40...120 кДж/моль, повышение температуры способствует хемосорбции в отличие от физической адсорбции. Примером такой адсорбции является адсорбция кислорода на вольфраме или кислорода на серебре при повышенных температурах.
При химической адсорбции молекулы адсорбата, связанные с адсорбентом прочными химическими силами, не могут перемещаться по поверхности последнего (локализованная адсорбция). В отличие от хемосорбции при физической адсорбции могут иметь место "как н е л о к а л и з о в а н н а я адсорбция, когда молекулы адсорбата способны передвигаться по поверхности адсорбента, так и локализованная адсорбция.
С повышением температуры локализованная физическая адсорбция может переходить в нелокализованную.
Как уже указывалось, физическая адсорбция протекает самопроизвольно, и этот процесс является динамическим: наряду с адсорбцией идет обратный процесс — десорбция, которая характеризуется удалением адсорбированных молекул с поверхности адсорбента. Скорость адсорбции с течением времени уменьшается, а скорость десорбции увеличивается. Эти изменения происходят до тех пор, пока обе скорости не становятся одинаковыми, т.е. не наступает адсорбционное равновесие:

 Адсорбция
Десорбция
Адсорбция
Десорбция
Для каждой температуры существует свое состояние равновесия. Чем выше концентрация адсорбата, тем больше адсорбция, а чем выше температура, тем меньше физическая адсорбция! Влияние температуры на физическую адсорбцию отвечает принципу Ле Шателье, поскольку десорбция как процесс, обратный адсорбции, сопровождается поглощением теплоты.
Так как химическая адсорбция обусловлена образованием связей, близких к химическим, десорбция протекает с большим трудом. При этом энергия активации возрастает с повышением степени покрытия поверхности хемосорбированными молекулами, что можно объяснить лишь существованием активных центров с различными энергиями активации.
Следует подчеркнуть, что явления физической и химической адсорбции четко различаются лишь в крайних случаях. Обычно осуществляются промежуточные варианты, когда основная масса адсорбированного вещества связывается сравнительно слабо (физическая адсорбция) и лишь небольшая часть связана прочно и может быть удалена длительным прогреванием и вакуумированием (химическая адсорбция). Например, кислород на металлах или водород на никеле адсорбируются при низких температурах по законам физической адсорбции, но при повышении температуры начинает протекать адсорбция с заметной энергией активации. В определенном интервале повышения температур прирост химической адсорбции перекрывает падение физической адсорбции, и на кривой температурной зависимости адсорбции возникает промежуточный максимум.
Адсорбционное равновесие является гетерогенным химическим равновесием. Поэтому каждому состоянию газа или пара с определенным давлением или температурой соответствует некоторое количество адсорбированного вещества, которое изменяется в соответствии с изменением параметров, определяющих состояние газа.
Уравнением адсорбции называют функциональную зависимость вида
f(Г, С, Т) = 0,
где Г — количество адсорбированного вещества (в кмолях), рассчитанное на 1 м2 поверхности адсорбента (или на 1 кг адсорбента); С — равновесная концентрация газа; Т — температура.
Это уравнение может быть представлено в виде
Г = f(C, Т),
определяющем зависимость количества адсорбированного газа от концентрации и температуры.
Если температура постоянная, то адсорбированное количество вещества есть функция исключительно равновесной концентрации газа:
Г = f(C); Т = const.
Это уравнение называется изотермой адсорбции. Часто изотермой адсорбции называют также графическую зависимость адсорбции от равновесного давления при постоянной температуре.
ТЕОРИИ АДСОРБЦИИ
В настоящее время нет общей теории, которая достаточно корректно описывала бы все виды адсорбции на различных адсорбентах и разных поверхностях раздела фаз. Поэтому рассмотрим наиболее распространенные теории, которые (несмотря на большое число допущений) позволяют на качественном уровне получить представление о таком сложнейшем процессе, как адсорбция.
Теория молекулярной адсорбции Ленгмюра. В основе этой теории лежат следующие положения.
1. Адсорбция является локализованной и вызывается силами, близкими к химическим.
2. Адсорбция молекул адсорбата происходит на активных центрах, всегда существующих на поверхности адсорбента. Такими центрами являются пики и возвышения, имеющиеся на любой поверхности. Так, например, на поверхности кристалла известкового шпата есть выступы высотой в 10-7...10-12 м, тонко отполированные зеркала имеют на поверхности выступы до 3 • 10-10 м.
Ряд исследователей считают также, что активными центрами
являются ребра и углы кристаллов и границы зерен в
микронеоднородном адсорбенте.
3. Активные центры характеризуются большой ненасыщенностью силового поля, благодаря чему на центрах удерживаются налетающие молекулы газа. Примером поверхности с центрами различной активности может служить поверхность восстановленного никеля. Чем больше свободных связей у атомов адсорбента или, иначе говоря, чем меньше данный атом связан с другими атомами, образующими поверхность адсорбента, тем сильнее осуществляется на них адсорбция.
4. Каждый активный центр обладает малым радиусом действия и способен насыщаться. Поэтому активный центр может провзаимодействовать лишь с одной молекулой адсорбата. В результате этого на поверхности адсорбента может образоваться только один (мономолекулярный) слой адсорбата (мономолекулярная адсорбция).
5. Адсорбированные молекулы удерживаются данным активным центром только в течение определенного промежутка времени. Через некоторое время молекулы отрываются от активного центра и переходят в газовую фазу. Взамен этих молекул активные центры могут адсорбировать новые молекулы, которые, в свою очередь, десорбируются, и т. д. Время пребывания молекулы в адсорбированном состоянии сильно зависит от температуры. При низких температурах это время может быть сколь угодно большим. При высоких температурах порядка 1300...2300 К время пребывания молекулы в адсорбированном состоянии может равняться всего 10-6 с.
6. Силы взаимодействия между адсорбированными молекулами не учитываются. Поэтому время пребывания молекул газа на активных центрах не зависит от того, заняты молекулами газа соседние активные центры или нет.
На основании приведенных положений было выведено уравнение изотермы локализованной адсорбции, пригодное как для описания адсорбции газов, так и растворенных веществ.
Представим себе однородную поверхность адсорбента, находящуюся при постоянной температуре в контакте с газом. Равновесие устанавливается тогда, когда число молекул адсорбата, остающихся на поверхности в единицу времени, становится равно числу молекул, покидающих поверхность за то же время. Число молекул, остающихся на адсорбенте, пропорционально концентрации С и доле поверхности, которая остается свободной. Если вся рассматриваемая поверхность имеет площадь, равную единице, и занята ее доля, равная , θ то доля (1 —θ) свободная. Следовательно, скорость адсорбции, т. е. скорость связывания молекул поверхностью,
__dθ__=R1C(1-θ)
dt
Скорость десорбции пропорциональна занятой поверхности:
__ __dθ__=R2θ
dt
При равновесии скорость адсорбции равна скорости десорбции. Поэтому, приравняв обе скорости, получим
R1C C
θ=-------------------- или θ=--------------- (11.1)
R2+R1C C+a
где
а = R2/R1.
Так как величина θ равна отношению адсорбированного количества вещества Г к максимальной адсорбции Гмакс при полном заполнении поверхности,
Г С
θ=---------==---------
Гмакс С+а
Отсюда
С
Г = Гмакс---------- (11.2)
С+а
где Г — адсорбция; С — равновесная концентрация газа; а — отношение скоростей десорбции и адсорбции.
Уравнение (11.2) называется уравнением изотермы адсорбции Ленгмюра. Оно показывает, что при малых концентрациях (при С « а) адсорбция пропорциональна концентрации:
Гмакс
Г=-------- С=КС
а
а при высоких концентрациях (С»а) адсорбция стремится к предельному значению Гмакс (рис. 93). При адсорбции газов С может быть заменено пропорциональной величиной давления газа. При адсорбции из раствора, для которой это уравнение, в принципе, также остается справедливым, С означает концентрацию растворенного вещества.
Уравнение Фрейндлиха. Представления, развитые И. Ленгмюром, в значительной степени идеализируют и упрощают действительную картину адсорбции. На самом деле поверхность большинства адсорбентов неоднородна, между адсорбированными частицами имеет место взаимодействие, и адсорбция часто не ограничивается образованием мономолекулярного слоя. В этом случае уравнение изотермы адсорбции усложняется. Г. Фрейндлих предположил, что масса адсорбированного газа или растворенного вещества, приходящаяся на единицу массы адсорбента, должна быть пропорциональна равновесному давлению (для газа) или равновесной концентрации (для твердого вещества, адсорбируемого из раствора), возведенной в какую-то дробную степень. Другими словами, чем выше давление и чем больше концентрация растворенного вещества, тем больше вещества будет адсорбции адсорбироваться на поверхности, Ленгмюра однако пропорциональность должна

Рис.93
носить не прямой, а экспоненциальный характер. Это положение выражается эмпирическим уравнением
х=Кр1/n или х=КС1/n (11.3)
где х — масса адсорбированного вещества, приходящаяся на 1 г адсорбирующего материала, г; р — равновесное давление; С — равновесная концентрация; К и n — константы.
Переписывая уравнение в логарифмической форме, получаем
1
lgх = ----(lgC)+lgК. (11.4)
n
Зависимость lgx от lgC выражается прямой линией, тангенс угла наклона которой равен 1/n, отсекающей на оси ординат отрезок, равный lgК
Предполагая экспоненциальное распределение активных центров по энергиям адсорбции, Я.Б.Зельдович теоретически получил уравнение, которое ранее эмпирически было установлено Г. Фрейндлихом.
На рис. 94 приведены изотермы адсорбции уксусной и бензойной кислот на древесном угле при 298 К, подчиняющиеся уравнению Фрейндлиха.
Теория полимолекулярной адсорбции. Эксперименты показали, что на практике, особенно при адсорбции паров, встречаются изотермы, правая часть которых круто поднимается кверху (рис. 95), что свидетельствует о взаимодействии адсорбированных слоев молекул с адсорбатом, когда адсорбированные молекулы наслаиваются друг на друга. Для объяснения этого явления и описания, так называемых S-образных изотерм адсорбции М. Поляни (1915) предложил теорию полимолекулярной адсорбции. В основе этой теории лежали следующие положения.
1. Адсорбция создается чисто физическими силами.
2. На поверхности адсорбента нет активных центров, адсорбционные силы действуют вблизи от поверхности адсорбента, образуя непрерывное силовое поле.
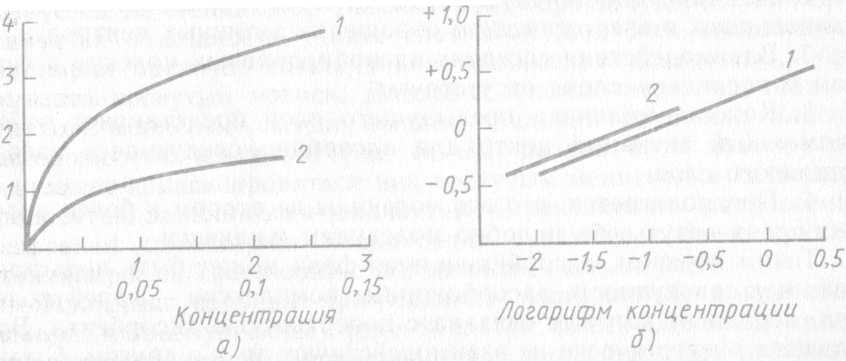
Рис 94.
Графики Фрейндлиха, построенные с использованием уравнений (11.3) (а) и (11.4) (б):
1-уксусная кислота в воде; 2 — бензойная кислота в бензоле
Рис.95Изотерма полимолекулярной адсорбции
3. Силовое поле, обусловливающее адсорбцию, действует на расстояния, которые больше, чем размеры отдельных молекул адсорбата^ Иначе говоря, у поверхности адсорбента существует так называемый адсорбционный объем, который заполняется при адсорбции молекулами адсорбата.
4.Притяжение молекулы адсорбции адсорбата поверхностью адсорбента не зависит от наличия в адсорбционном объеме других молекул, вследствие чего возможна полимолекулярная адсорбция.
5. Адсорбционные силы не зависят от температуры и, следовательно, с изменением температуры адсорбционный объем не изменяется.
Обе рассмотренные теории адсорбции — теория мономолекулярной адсорбции Ленгмюра и теория полимолекулярной адсорбции не могут описать с достаточной полнотой сложного процесса адсорбции.
Обобщенная теория Брунауэра, Эммета и Теллера. Делались попытки обобщить представления И. Ленгмюра и М. Поляни и описать изотермы различной формы с помощью одного уравнения. Такая обобщенная теория была развита С. Брунауэром, П. Эмметом и Е. Теллером (1935... 1940) применительно к адсорбции паров. Она получила название БЭТ по первым буквам фамилий авторов. Основные положения этой теории следующие.
1. На поверхности адсорбента имеется определенное число равноценных в энергетическом отношении активных центров.
2. Взаимодействия соседних адсорбированных молекул в первом и последнем слоях отсутствуют.
3. Каждая молекула предыдущего слоя представляет собой возможный активный центр для адсорбции следующего адсорбционного слоя.
4. Предполагается, что все молекулы во втором и более далеких слоях ведут себя подобно молекулам жидкости.
Таким образом, адсорбированная фаза может быть представлена как совокупность адсорбционных комплексов — цепей молекул, первая из которых связана с поверхностью адсорбента. Все эти цепи энергетически не взаимодействуют друг с другом. Схема строения адсорбционного слоя по теории БЭТ показана на рис. 96.
С. Брунауэр, П. Эммет и Е. Теллер вывели уравнение
![]()
Рис. 96. Схема строения адсорбционного слоя по теории БЭТ
___Р___=__1__+___С-1 _Р__
V(po-p) VмC VмC ро (11.5)
где ро — давление насыщенного пара; V— объем адсорбированного газа при данном давлении газа р; VM — объем адсорбированного газа в монослое; С = еΔε/RT (Δε — разность между теплотой адсорбции газа в первом слое и теплотой сжижения газа).
Находясь зависимость левой части уравнения (11.5) от р/ро, получают на графике прямую линию, наклон которой равен С — 1/(VмС), а отрезок на оси ординат 1/(VMC). Отсюда можно определить величины VM и С.
Из значения VM может быть определена полная адсорбирующая поверхность адсорбента S, что имеет важное практическое значение, а из значения С вычисляется теплота адсорбции газа в монослое (обычно около 8... 10 кДж/моль). Например, адсорбирующая поверхность 1 кг силикагеля составляет 5-10 м2/кг, а активированного угля — до 1-106 м2/кг.
Капиллярная конденсация. При сорбции паров на твердых пористых адсорбентах адсорбционный процесс может перейти в так называемую капиллярную конденсацию. Сначала пар адсорбируется на стенках пор (капилляров) сорбента, а затем конденсируется в жидкость. Далее слои этой жидкости соединяются, заполняя частично самые тонкие капилляры жидкостью, образующей вогнутый мениск. Давление насыщенного пара над вогнутым мениском всегда меньше давления пара над плоской поверхностью жидкости и поэтому пар начинает конденсироваться над вогнутым мениском и капилляры полностью заполняются жидкостью. С увеличением равновесного давления капиллярная конденсация охватывает более крупные капилляры по сравнению с первичными капиллярами.
Изотерма сорбции при наличии капиллярной конденсации имеет S-образную форму. При малых давлениях кривая представляет собой обычную изотерму адсорбции, при приближении к пределу адсорбции она резко поднимается вверх. Этот участок соответствует массе сорбированного вещества в результате капиллярной конденсации.
4. ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ. СМАЧИВАНИЕ
Гетерогенная система, представляющая собой чистую жидкость, которая соприкасается с другой средой, например с ее собственным паром, характеризуется тем, что жидкость на поверхности находится в особых условиях по сравнению с остальной массой жидкости.
Каждая молекула во внутренних слоях жидкости подвергается
одинаковому воздействию других молекул, окружающих ее со всех
сторон. Поэтому силы молекулярного воздействия здесь уравновешиваются.
Если же молекула жидкости находится в поверхностном слое на границе с газом, то взаимодействие этой молекулы с молекулами газа намного меньше, чем с молекулами жидкости. В результате этого молекулы на границе с газом стремятся втянуться внутрь жидкости, а это значит, что поверхность раздела жидкость — газ стремится к уменьшению. Этот процесс обусловлен известным термодинамическим стремлением свободной энергии к уменьшению. Наиболее ярким проявлением этого процесса может служить шарообразная форма свободной капли жидкости; известно, что шар из всех геометрических фигур данного объема обладает минимальной поверхностью.
Итак, если известно, что молекулы из поверхности раздела самопроизвольно стремятся внутрь, то ясно, что обратный процесс — перемещение молекулы из объема на поверхность — потребует затраты энергии. Увеличение поверхности жидкости требует затраты энергии F, равной работе Ó1, необходимой для перемещения одной молекулы из объема на поверхность, умноженной на число перемещенных молекул n:
F= n σ1.
Если работа проводится в равновесных изотермических условиях, то величина F, входящая в это уравнение, называется свободной поверхностной энергией или поверхностной энергией, которая, отнесенная к единице поверхности, называется поверхностным натяжением:
σ= F/S,
где σ— поверхностное натяжение, Дж/м2; F — поверхностная энергия; S — площадь поверхности.
Поверхностное натяжение для различных жидкостей неодинаково и особенно велико у воды.
В табл. 43 приведены значения поверхностного натяжения для некоторых веществ.
Вещество |
Газовая атмосфера |
Т, К |
σ , Дж/м2 |
Бром |
Воздух |
286 |
44,1 |
Сера |
» |
411 |
58,3 |
Цинк |
Вакуум |
700 |
772,2 |
Ртуть |
» |
293 |
471,6 |
Вода |
Воздух+П |
373 |
58,80 |
Ацетон |
Воздух |
293 |
23,7 |
Метанол |
» |
288 |
22,99 |
Масляная кислота |
П |
288 |
27,32 |
Октан |
Воздух |
293 |
31,11 |
Бензол |
» |
293 |
28,88 |
Толуол |
» |
293 |
27,4 |
Нафталин
|
» |
353 |
32,06 |
Фенол |
П |
328 |
36,5 |
Анилин |
» |
292,5 |
43,3 |
Поверхностное натяжение жидкости σ— это сила, приложенная по касательной к плоскости поверхности (приходящаяся на единицу длины), которая препятствует увеличению поверхности. Поверхностное натяжение, можно вычислить по уравнению:
σ =F/2L
где L — длина проволоки; множитель 2 учитывает наличие у жидкой пленки двух поверхностей — передней и задней.
Поверхностное натяжение чаще вычисляют, зная массу капли Жидкости в момент отрыва ее от капилляра, по уравнению
σ = mg/(2πr),
где г — радиус капилляра; σ — поверхностное натяжение; g— ускорение свободного падения.
Массу одной капли находят по уравнению
m = V Q /n,
где n — число капель в объеме V жидкости, вытекающей из капилляра; Q — плотность жидкости. Отсюда
σ = VQ g/(2πrn).
Если две жидкости, занимающие одинаковые объемы, поочередно пропускать через один и тот же капилляр, то
__ σ 1___=__Q1n2___
σ2 Q2n1
откуда находим
Q2 n2
σ1 = σ2 -----------
Q2 n1
Для определения поверхностного натяжения через установленный вертикально капилляр пропускают определенный объем исследуемой жидкости и считают капли, отрывающиеся от капилляра. Затем пропускают через капилляр такой же объем жидкости с известным поверхностным натяжением, обычно воду. Зная число капель n1и n2 в объеме V для двух жидкостей и их плотность, вычисляют поверхностное натяжение исследуемой жидкости.
Поверхностное натяжение на границе двух несмешивающихся жидкостей называется межфазным поверхностным натяжением. Чем меньше поверхностное натяжение на границе двух жидкостей (σ a,b), тем выше взаимная растворимость жидкостей и при критической температуре полного смешения обоих жидкостей (например, для системы вода — фенол при 339К)
σ a,b = 0.
По правилу Антонова межфазное поверхностное натяжение двух жидкостей, находящихся во взаимном равновесии, связано с поверхностным натяжением каждой из них соотношением
σ a,b = σ a — σ b
где σ a,b — поверхностное натяжение на поверхности раздела двух жидкостей; σ a и σ b — соответственно поверхностные натяжения жидкостей а и b на поверхности раздела жидкость/пар.
Растекание одной жидкости по поверхности другой. Гаркинс и его сотрудники подробно рассмотрели термодинамику образования новой поверхности, возникающей при соприкосновении двух жидкостей. Если жидкость В растекается изотермически и обратимо на поверхности жидкости А, то поверхность а исчезает и вместо нее появляется поверхность b; кроме этого, образуется поверхность раздела ab. Уменьшение свободной энергии ΔG, сопровождающее растекание, дается выражением
ΔG = σ a — σ a,b — σ b,
так как
АА = σ a — σ a,b + σ b , Ак = 2 σ b,
то
ΔG = АА -AK,
где АА—работа прилипания (адгезии); Ак — работа сцепления (когезии) жидкости.
Рассмотрим подробнее, что из себя представляют адгезия и когезия. Если имеется система, состоящая из молекул однородной жидкости (например, вода), то для создания новых поверхностей раздела (например, вода — воздух) потребуется затрата работы. Эта работа, затраченная на преодоление сил сцепления между молекулами однородной жидкости, называется к о г е з и е й.
Предположим, что имеется система, состоящая из двух неоднородных жидкостей с некоторой поверхностью раздела между ними (например, масло — вода). Если теперь создать новые поверхности раздела (например, масло — воздух и вода — воздух), то работа, связанная с «разрезанием» по имеющейся ранее поверхности раздела (масло — вода), будет затрачена на преодоление сил сцепления между разными молекулами. Такая работа называется адгезией.
Согласно правилу, установленному Гаркинсом, растекание происходит, если прилипание между двумя жидкостями больше, чем сцепление растекающейся жидкости. Следовательно, при АА — Ак > 0 происходит растекание, если же АА — Ак < 0, растекание не имеет места.
Следует заметить, что если жидкость а растекается на поверхности жидкости b, то это не значит, что жидкость b будет обязательно растекаться по поверхности жидкости а. Например, почти все органические жидкости растекаются по поверхности воды, но вода растекается по поверхности очень немногих органических веществ.
Поверхностное натяжение на границе трех фаз. На границе раздела трех фаз наблюдаются более сложные соотношения между межфазными поверхностными натяжениями. Если на твердую поверхность 3 (рис. 103)
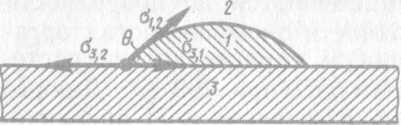
Рис. 103. Смачивание на границе раздела трех фаз (по Ребиндеру): 1 — вода; 2 — среда; 3 — твердая поверхность
нанесена капля воды 1 и обе поверхности граничат с газом 2, то капля образует с твердой поверхностью краевой угол смачивания θ (измеряемый в водной фазе). По уравнению Лапласа величина cos θ при равновесии связана с межфазными поверхностными натяжениями следующим соотношением:
σ3,2- σ3,1
cos θ=----------------------
σ1,2
где индексы σ указывают границы раздела фаз.
В зависимости от значений равновесного краевого угла различают три основных вида смачивания:
1) несмачивание (плохое смачивание) — краевой угол тупой: 180° > θ > 90°. Пример: вода на парафине или тефлоне;
2) смачивание (ограниченное смачивание) — краевой угол острый: 90° > θ>0°. Например, вода на металле, покрытом оксидной пленкой;
3) полное смачивание. Равновесный краевой угол не устанавливается, капля растекается в тонкую пленку. Например, ртуть на поверхности свинца, очищенной от оксидной пленки.
Значение равновесного краевого угла определяется соотношением сил притяжения жидкости к твердому телу (или к жидкой подложке) и сил взаимного притяжения между частицами (молекулами) самой жидкости. Эту зависимость можно продемонстрировать с помощью очень простых и наглядных опытов.
Рассмотрим смачивание при контакте воды с тщательно обезжиренным стеклом. Если стеклянную пластину погрузить в воду, а затем снова вытащить на воздух, на стекле остается тонкий слой воды. Это означает, что силы притяжения жидкости к твердому телу преобладают над взаимным притяжением молекул жидкости. Напротив, при отсутствии смачивания (например, при контакте ртути со стеклом) после аналогичного опыта на твердой поверхности не остается следов жидкости.
Связь между молекулярным взаимодействием жидкости и твердого тела и видом смачивания выявляется особенно отчетливо при избирательном смачивании, когда с твердым телом одновременно контактируют две жидкости, различные по своей молекулярной природе (полярная и неполярная). По виду избирательного смачивания все твердые тела подразделяют на три основные группу:
1) гидрофильные (или олеофобные) материалы, которые лучше смачиваются водой: кальцит, кварц, большинство силикатов и окисленных минералов, галогениды щелочных металлов;
2) гидрофобные (олеофильные) материалы, которые лучше смачиваются неполярной жидкостью (мылом): графит, уголь, сера;
3) абсолютно гидрофобные тела; в эту группу входят парафин, тефлон, битумы.
Помимо краевого угла смачивания, другой мерой гидрофильноcсти поверхности является теплота смачивания, так как гидрофильные поверхности смачиваются водой с положительным тепловым эффектом. По П. А. Ребиндеру, гидрофильность поверхности следует характеризовать по отношению теплот ее смачивания водой q1 и бензолом q2. Для гидрофильной поверхности
q1
------- >1 (для агара 35),
q2
для гидрофобной поверхности
q1
--------- < 1 (для угля 0,34).
q2
Капиллярное давление. В различных процессах, связанных со смачиванием, важную роль играет капиллярное давление, которое возникает из-за искривления поверхности жидкости. Для выпуклой поверхности капиллярное давление положительно, для вогнутой оно отрицательно. Если поверхность раздела жидкости выпуклая (рис. 104),
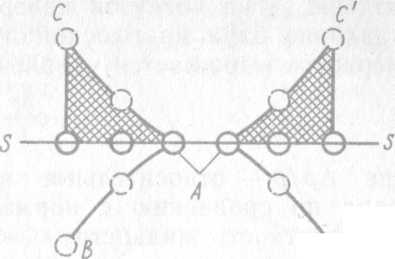
Рис. 104. Выпуклая (ВАВ'), плоская (SAS') и вогнутая (САС') поверхности жидкости.
то молекулы из поверхности жидкости (например, молекулы А) втягиваются внутрь жидкости меньшим числом молекул, т. е. слабее, чем из плоской поверхности (SAS'), а из вогнутой поверхности (САС') — большим числом молекул жидкости, т. е. сильнее, чем из плоской поверхности. Поэтому
между искривленной и плоской поверхностью раздела существует разность молекулярных давлений, которая называется капиллярным давлением р. Искривление поверхности характеризуют радиусом кривизны r, направленным внутрь жидкости при выпуклой поверхности (в этом случае r считается положительным) и наружу при вогнутой поверхности ( r отрицателен). Для плоской поверхности r = ∞. По уравнению Лапласа
р = 2 σ /r,
откуда видно, что для плоской поверхности р = 0, для выпуклой поверхности р>0 и для вогнутой поверхности р < 0; эти давления р суммируются с внешним давлением, оказываемым на жидкость.
Давление пара жидкости на выпуклой поверхности больше, а на вогнутой поверхности — меньше, чем нормальное давление пара на плоской поверхности жидкости. Эта закономерность выражается уравнением Томсона
Δp 2M
-------- = ----------- (11.9)
P RT Q r
Где Δ р/р — относительное изменение давления насыщенного пара по сравнению с нормальным; М — молекулярная масса; Q — плотность жидкости; r — радиус капилляра или капли жидкости.
Из уравнения (11.9) следует, например, что для капель воды с r = 10 -8 упругость пара на 10% выше, чем для воды с плоской поверхностью; поэтому если в замкнутой системе содержатся одновременно крупные и мелкие капли, последние перегоняются к крупным каплям. Уравнение (11.9) объясняет также более высокую растворимость мелких твердых частиц по сравнению с крупными, и ряд других явлений.
КОЛЛОИДНЫЕ СИСТЕМЫ И МЕТОДЫ ПОЛУЧЕНИЯ ЛИОФОБНЫХ КОЛЛОИДОВ
ОБЩАЯ ХАРАКТЕРИСТИКА КОЛЛОИДНЫХ СИСТЕМ
Возникновение коллоидной химии как самостоятельной науки связывают обычно с именем английского ученого Т. Грэма, который в 1861 г. разработал ряд методов приготовления и очистки коллоидных растворов. Т. Грэм предложил все вещества классифицировать на две группы:
1) кристаллоиды — сравнительно быстро диффундирующие вещества, образующие истинные растворы, из которых вещество при определенных условиях может быть выделено в виде кристаллов;
2) коллоиды — вещества, обладающие малой скоростью диффузии, низким осмотическим давлением и образующие коллоидные растворы, студни или клееподобные аморфные осадки.
Т. Грэм ошибочно считал, что коллоиды и кристаллоиды представляют собой совершенно обособленные классы веществ, между которыми нет никакого сходства. На основе теоретических соображений и тщательного экспериментального исследования русские ученые Н. Г. Борщев (1869) и П. П. Веймарн (1904...1916) сделали правильный вывод о том, что любое вещество можно перевести в коллоидное состояние, создавая соответствующие условия.
Если коллоидные частицы находятся в жидкой среде, то такие системы обычно называют коллоидными растворами или золями.
В течение XIX и в начале XX вв. был выполнен ряд фундаментальных исследований, сыгравших большую роль в развитии коллоидной химии. На основе этих исследований было установлено, что в коллоидных растворах частицы находятся в высокой степени раздробления или диспергирования, но они гораздо больше молекул.
Таким образом, коллоидные системы относятся к дисперсным системам, т. е. к системам, где одно вещество в виде частиц различной величины распределено в другом веществе.
Степень раздробленности частиц называется степенью д и с п е р с н о с т и. Среда, в которой находится раздробленное (диспергированное) вещество, называется дисперсионной средой, а раздробленное вещество в виде частиц разных размеров — д и с п е р с н о й ф а з о й.
Дисперсные системы чрезвычайно многообразны и можно сказать, что они представляют основу всего биологического мира. Распространенность их в технике также очень велика, так как различие химического состава отдельных компонентов дисперсных систем, величины частиц и разнообразие дисперсионных сред обеспечивают получение и использование колоссального числа продуктов.
Дисперсионные системы прежде всего классифицируют по размеру частиц дисперсионной фазы или, иначе говоря, по степени дисперсности (табл. 45). Кроме того, их разделяют на группы, отличающиеся по природе и агрегатному состоянию дисперсной фазы и дисперсионной среды.
Таблица 45. Дисперсные системы
Системы |
Диаметр частицы, м |
Грубодисперсные системы |
10-7... 10-5 |
Коллоидные системы |
10-9...10-7 |
Истинные растворы |
<10-9 |
Грубодисперсные системы. Если дисперсная фаза состоит из твердых частиц, то система называется в з в е с ь ю или с у с п е н з и е й. В качестве примера можно привести взмученную глину в воде. Если дисперсная фаза представлена капельками жидкости, то ее называют э м у л ь с и е й; пример эмульсии — капли масла в воде. Эти системы неустойчивы.
Коллоидные системы. Частицы коллоидных систем значительно больше молекул (ионов), из которых состоит дисперсионная среда, что приводит к наличию поверхности раздела между частицами и средой. Если суспензии можно наблюдать с помощью микроскопа, то коллоидные частицы таким способом не обнаруживаются.
Коллоидные системы относительно устойчивы, но со временем они разрушаются. При получении коллоидных систем затрачивается внешняя энергия.
Истинные растворы. Их иначе называют молекулярно-дисперсными или ионно-дисперсными системами. Эти растворы устойчивы, не разрушаются и получаются самопроизвольно.
Исследования коллоидных систем, особенно изучение зависимости их устойчивости от наличия и концентрации электролитов в растворе, показали, что одна лишь дисперсность не может охарактеризовать коллоидные системы и объяснить их свойства.
Н. П. Песков (1917) установил, что свойства коллоидных систем зависят не только от размеров частиц, но и в гораздо большей мере от наличия поверхностей раздела со значительной свободной поверхностной энергией. Был сделан вывод, что кроме кинетической устойчивости, зависящей от размера частиц, имеется устойчивость частиц к взаимному слипанию (агрегативная устойчивость).
Коллоидным системам свойственна агрегативная неустойчивость, преодолеваемая лишь путем адсорбции ионов или молекул на частицах дисперсной фазы. Таким образом, агрегативно-устойчивая коллоидная система, в принципе, должна состоять из трех компонентов: диспергированных частиц, среды и стабилизатора.
Глубокое изучение процессов и характера взаимодействия дисперсной фазы и дисперсионной среды привело к тому, что коллоидные системы не могут рассматриваться как единое целое, а имеются две группы, резко отличные по взаимодействиям между частицами дисперсной фазы и дисперсионной среды: л и о ф о б н ы е к о л л о и д ы и р а с т в о р ы в ы с о к о м о л е к у л я р н ы х соединений (растворы ВМС), которые ранее назывались лиофильными коллоидами.
К лиофобным коллоидам относятся системы, в которой частицы дисперсной фазы не взаимодействуют или слабо взаимодействуют с дисперсионной средой. Эти растворы получают с затратой энергии, и они устойчивы лишь в присутствии стабилизаторов.
К растворам ВМС относятся системы, образованные самопроизвольно, благодаря сильному взаимодействию дисперсной фазы и дисперсионной среды.
Работами В. А. Каргина и его школы было показано, что растворы ВМС являются термодинамически обратимыми молекулярными гомогенными (однофазными) системами, способными сохранять агрегативную устойчивость без стабилизатора в двухкомпонентном растворе.
Лиофобные коллоиды и растворы ВМС отличаются также и по структуре частиц, составляющих дисперсную фазу. Для лиофобных частиц характерной единицей структуры является м и ц е л л а, представляющая собой сложный многокомпонентный агрегат с переменным числом адсорбированных ионов или молекул. Растворы ВМС представляют собой истинные растворы.
Осмотическое давление
Уравнение Вант-Гоффа применимо лишь к идеальным или разбавленным растворам. Однако это уравнение с некоторыми оговорками может быть использовано и при рассмотрении коллоидных растворов.
Прежде всего следует отметить, что значение осмотического давления пропорционально числу частиц растворенного вещества в растворе. Поэтому при переходе от истинных растворов к коллоидным, где размер частиц намного больше (а значит, число частиц при той же концентрации намного меньше), произойдет резкое изменение давления. Так, можно показать, что если в 1 М истинном растворе осмотическое давление равно 22,5 кПа, то для 0,1%-ного золя золота с размером частиц 10-8 м осмотическое давление будет приблизительно равно 22,5-10-7 кПа, т. е. в 107 раз меньше. Столь малое значение не только нельзя использовать для проверки применимости теории к коллоидным растворам, но ее почти невозможно измерить с необходимой точностью, учитывая погрешности осмотических экспериментов.
Таким образом, в процессе осмоса не обнаруживается никаких принципиальных качественных различий между коллоидными и молекулярными растворами (основные закономерности едины), но большие количественные различия в значениях концентрации приводят к слабому проявлению осмоса в коллоидных системах.
Размер частиц в коллоидных растворах может колебаться в довольно широких пределах, поэтому для различных коллоидных растворов значения осмотического давления будут резко отличаться, что связано с разной дисперсностью коллоидных систем. Простой расчет позволяет установить количественную зависимость между осмотическим давлением, числом частиц n в единице объема раствора, средним радиусом r частиц, а значит, и степенью дисперсности z.
Так, массу диспергированного вещества в единице объема раствора можно выразить через 4/3πr3dn (где d — плотность раствора). Тогда для двух дисперсных систем с одинаковым растворителем и при одинаковых температурных условиях можно записать
4/3πr31dn1=4/3πr32dn2
И
r31n1=r32n2
Таким образом,
n1 p1 r32 z31
------=-------=-------=---------
n2 p2 r31 z32
т. е. осмотическое давление обратно пропорционально кубу радиуса частиц и, следовательно, прямо пропорционально кубу степени дисперсности.
Это значит, что даже самое небольшое изменение в размерах частиц дисперсных систем должно повлечь за собой весьма значительные изменения в осмотическом давлении. Например, при увеличении размера частиц в 2 раза осмотическое давление должно уменьшиться в 8 раз при той же концентрации раствора.
Растворы лиофобных коллоидов характеризуются непостоянным размером частиц. При увеличении концентрации, например, возможно увеличение размеров этих частиц благодаря процессу агрегации (слипания), а при уменьшении концентрации — процессу дезагрегации (разукрупнения). Эти процессы приводят к тому, что с изменением концентрации осмотическое давление изменяется не прямо пропорционально последней, как следует из уравнения Вант-Гоффа, а значительно больше или меньше.
Так как на процессы укрупнения и разукрупнения частиц определенное влияние оказывает и температура, осмотическое давление с ростом температуры изменяется для ряда коллоидных систем аномально:
с повышением температуры осмотическое давление растет не по прямой в соответствии с уравнением Вант-Гоффа,
а по быстро восходящей кривой и с понижением оно аномально падает и притом по иной, нисходящей, кривой.