

Раздел II. Общие закономерности химических процессов. Химическая динамика тема 8. Химическая кинетика и катализ
§1. Скорость химической реакции, ее зависимость от
различных факторов. Реакции I-го и II-го порядка
Законы термодинамики позволяют определить потенциальные возможности любого химического процесса, но ничего не говорят о его природе, характере, механизме и скорости.
Раздел химии, изучающий количественные закономерности и механизмы химических превращений, называют химической кинетикой.
Главное предназначение кинетики - обеспечение возможности управлять процессами, создавать продукты с заданными свойствами, предотвращать нежелательные побочные процессы.
Наряду со свободной энергией (ΔG и ΔF) количественной характеристикой потенциальных возможностей химического процесса является его скорость.
Скоростью химического процесса называют изменение количества вещества реакционной системы в единицу времени в единице объема реакционного пространства. Таким образом определенная скорость обозначается буквой υ или с и называется средней скоростью процесса за определенный промежуток времени.
В зависимости от природы химического процесса меняется и характер реакционного пространства. Так, если химический процесс совершается в гомогенной среде (и реагенты и продукты находятся в одном агрегатном состоянии), реакционным пространством для него служит объем сосуда, заполненный реагентами. В этом случае:
Средняя скорость гомогенной химической реакции определяется по изменению концентрации любого из реагентов или продуктов в
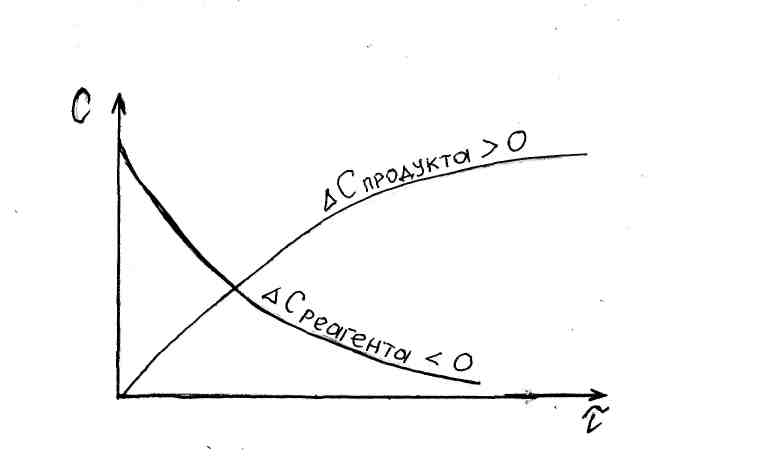
единицу времени; υi = ±ΔСi/Δτ. На рис.52 представлено изменение концентрации реагентов и продуктов во время совершения химической реакции.
Как видим, ΔСiреаг= Сi2 – Сi1<0, а ΔСiпрод = Сi2 – Ci1>0, скорость же процесса по определению не может быть отрицательной величиной, вот почему при мониторинге скорости по реагентам перед дробью ставится знак
минус «–».
Однако, средняя скорость не позволяет достаточно быстро реагировать на те или иные изменения во время химического процесса, особенно если эта
скорость высока.
за определенный промежуток времени.
Для мониторинга непрерывных процессов необходимо знать величину скорости в любой конкретный момент времени, т.е. определять мгновенную или истинную скорость процесса, а не среднюю.
Истинная скорость химического процесса характеризуется изменением концентрации любого компонента химической реакции (продукта или реагента) в данный конкретный момент времени.
С математической точки зрения это означает, что истинная скорость химического процесса по компоненту i (υi) будет рассчитываться как предел средней скорости при Δτ→0, т.е. υi = lim(±ΔСi/Δτ) = ±dСi/dτ.
Уравнение υi = ±dСi/dτ или υ = ±dС/dτ называется кинетическим уравнением для любого химического процесса. Скорость как физическая величина измеряется в [моль/л·с].
На скорость химического процесса влияет очень много различных факторов: природа реагентов и характер взаимодействия между ними (одни реакции идут мгновенно и со взрывом, а другие медленно, практически незаметно и тянутся десятками и сотнями лет), концентрация вступающих в реакцию исходных веществ, температура процесса, внешнее воздействие (например, излучение или пластическая деформация), присутствие катализатора, и даже иногда форма сосуда, в котором совершается реакция.
Влияние концентрации участников реакции на скорость процесса определяется экспериментальным путем. Взаимодействие между частицами реагентов происходит лишь в момент их реального физического столкновения, причем, с силой, достаточной для перехода кинетической энергии движущихся частиц в потенциальную энергию взаимодействия между ними. Если температура реакционной среды не изменяется, то число столкновений между частицами будет зависеть от количества самих частиц (чем их больше, тем больше вероятность эффективных соударений между ними). Следовательно, увеличение концентрации компонентов системы увеличивает скорость химического процесса.
Эта зависимость была установлена эмпирическим путем в 1864-1867гг норвежскими химиками К. Гульдбергом и П. Вааге и носит название основного постулата химической кинетики или закона действующих масс для кинетики.
Прежде чем мы сформулируем этот закон, запишем в виде схемы в общем виде уравнение реакции практически необратимого химического процесса (теоретически, как мы помним, все процессы являются обратимыми):
аА + bВ + dD + … = mM + nN + ℓL + … В этом уравнении обозначим молярные концентрации реагентов, возведенные в степени, соответствующие коэффициентам, с которыми эти реагенты входят в уравнение химической реакции, как Сμа(А); Сμb(В); Сμd(D). Такие величины в кинетике называют действующими массами реагентов.
Теперь сформулируем сам закон действующих масс:
Скорость практически необратимой химической реакции находится в прямой пропорциональной зависимости от действующих масс ее реагентов.
Математическое выражение закона действующих масс имеет вид:
υ = k·Сμа(А)·Сμb(В)·Сμd(D)…
Коэффициент пропорциональности (k) в законе действующих масс имеет физический смысл и называется константой скорости химической реакции. Легко установить, что при Сμ = 1моль/л скорость процесса υ = k, следовательно, «k» не зависит от концентрации реагентов, но зависит от их природы и от температуры, поэтому константа скорости является главной характеристикой любого химического процесса.
Закон действующих масс в представленном виде носит обобщенный характер. Показатели степени, в которые возведены концентрации реагентов (a,b,d…) называют «порядком реакции по реагенту A, B, D …», т. е.:
a = nA – порядок реакции по реагенту А;
b = nB – порядок реакции по реагенту В;
d = nD – порядок реакции по реагенту D и т.д.
Сумма порядков реакции по реагентам определяет общий порядок конкретной химической реакции (n): n = nA + nB + nD + …= ∑ni , где ni – порядок реакции по i – тому компоненту.
Например, для реакции 2СО + О2 = 2СО2 скорость химической реакции (υ), согласно закону действующих масс, выразится следующим уравнением: υ = k·Сμ2(СО)·Сμ(О2), а порядок этой реакции (n) будет равен n=2+1=3. Если процесс идет при эквивалентных соотношениях реагентов и Сμ (СО) = Сμ (О2), то кинетическое уравнение примет вид
υ = kСμ3, что прямо указывает на его порядок.
Для реакции N2 + O2 = 2NO скорость определится как
υ = k·Сμ(N2)·Cμ(O2) или υ = К·Сμ2, что указывает на 2-й порядок. А вот реакция разложения кислорода на атомарный кислород О2 = 2О˙ является реакцией первого порядка. Уравнение υ = f (Cn) называют уравнением формальной кинетики и по нему эмпирическим путем определяют скорость для реакций 1-го, 2-го, …, n –го порядка.
Так, скорость реакции 1-го порядка подчиняется уравнению υ=kСμ (по закону действующих масс) и уравнению υ= – dCμ/dτ (по определению понятия скорости). Приравнивая эти два уравнения, получим: dCμ/dτ = – kСμ
или, после разделения переменных, dCμ/Сμ = – kdτ. Решение полученного дифференциального уравнения при первоначальных условиях, что τ0=0 и Сμ при τ0 равна Сμ=C0, приведет к выражению: ∫dCμ/Сμ =∫ – kdτ, а далее при
взятии интеграла Сμ=C0·e-kτ . Подставив полученное значение концентрации реагента Сμ в закон действующих масс, получим окончательное выражение для скорости реакции 1-го порядка: υІ=kІ ·C0 ·e–kτ .
Если дифференциальное уравнение решать относительно константы скорости реакции k, то после его логарифмирования получим
ℓ nCμ
= ℓnCo
– kτ
и, следовательно, kI
= τ-1·ℓnCo/Cμ
. На рис. 53 показан график зависимости
ℓnCμ
от τ , представляющий собой прямую
линию. Отрезок, отсекаемый прямой на
оси ординат, равен ℓnCo
, а тангенс угла наклона прямой к оси
абсцисс дает возможность определить
константу скорости реакции первого
порядка kI
= tgα
.
nCμ
= ℓnCo
– kτ
и, следовательно, kI
= τ-1·ℓnCo/Cμ
. На рис. 53 показан график зависимости
ℓnCμ
от τ , представляющий собой прямую
линию. Отрезок, отсекаемый прямой на
оси ординат, равен ℓnCo
, а тангенс угла наклона прямой к оси
абсцисс дает возможность определить
константу скорости реакции первого
порядка kI
= tgα
.
Большинство ядерных реакций являются реакциями 1-го порядка и для них характерно использование параметра «период полураспада τ½» - это время, в течение которого концентрация реагента уменьшается вдвое по сравнению с начальной концентрацией.
Для расчета τ½ в выражение для константы скорости реакции первого порядка подставим вместо τ – τ½ , а вместо Сμ – Со/2. В результате получим:
kI = (1/τ½)·ℓn2 , или τ½I= ℓn2/kI = 0,693/kI .
Как видим, период полураспада для реакций I-го порядка не зависит от концентрации.
Скорость реакции 2-го порядка для двухкомпонентной системы А + В = АВ подчиняется кинетическому уравнению υІІ=kІІ·Cμ(A)·Cμ(B). Если принять, что в этом уравнении Cμ(A) = Cμ(B), то υІІ=kІІ·Cμ2 . В то же время, согласно основному постулату химической кинетики υ= -dCμ/dτ.
Объединим оба уравнения для скорости реакции в систему и, разделив переменные, получим дифференциальное уравнение dCμ/ Cμ2 = –kІІ·dτ. Интегрируем его при условии, что время изменяется от τ0=0 до τ , а концентрация – от C0 до Cμ: ∫dCμ/ Cμ2 =∫ –kІІ·dτ . При решении получим Cμ=C0/(1+kII·Co·τ) . Это значение концентрации реагентов подставим в уравнение для скорости реакции 2-го порядка и решим его относительно υІІ и относительно kІІ. В результате получим уравнения, с помощью которых можно произвести расчеты скорости и константы скорости реакции 2-го порядка:
Cо2 1 1 1 (Со-Сμ)
υІІ=kІІ·——————; kІІ= —·(— – —)=-------------- ·
(1+kІІ·Cо·τ)2 τ Cμ Cо τCμCо

от времени протекания реакции 1-го порядка.
Если построить график зависимости концентрации реагента от времени реакции 2-го порядка в координатах (1/Сμ ÷τ), то он также будет представлять из себя прямую линию, отсекающую на оси ординат отрезок, равный 1/С0, а по тангенсу угла наклона прямой к оси абсцисс можно определить константу скорости реакции II-го порядка: k = tgα ( см. рис. 54).
 Период
полураспада для реакций второго порядка
определяется по уравнению: τ½ІІ=1/kІІ·C0.
Период
полураспада для реакций второго порядка
определяется по уравнению: τ½ІІ=1/kІІ·C0.
Все приведенные нами кинетические уравнения относились к гомогенным системам. Гетерогенные реакции протекают на поверхности раздела фаз, именно поверхность и служит реакционным пространством в этих случаях. Тогда закон действующих масс будет звучать несколько иначе:
Скорость гетерогенной химической реакции находится в прямой пропорциональной зависимости от концентрации (действующей массы) неконденсированной (наименее конденсированной) фазы реагентов и площади реакционной поверхности (площади поверхности
конденсированной фазы).
Р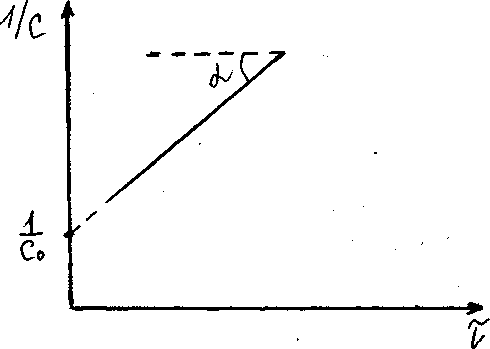 ис.54.
Зависимость скорости реакции второго
порядка от времени процесса.
ис.54.
Зависимость скорости реакции второго
порядка от времени процесса.
Следовательно, для гетерогенной реакции I-го порядка выражение скорости будет иметь вид: υІ=kІ·Cμ·S, где Сμ – концентрация газообразного реагента, S – площадь поверхности твердого реагента.
Например, скорость гетерогенной реакции СаО(т) + СО2(г) = СаСО3(т) определится по формуле υІ=kІ·Cμ(СО2)·S(СаО). Как следует из приведенной формулы, чем больше площадь поверхности твердого реагента, тем выше скорость гетерогенной реакции. Вот почему требуется измельчение твердой фазы для ускорения процесса, либо наращивание пористого покрытия, увеличивающего количество активных центров реакционного пространства.
Если реакция происходит между твердыми веществами, то ее скорость вообще не будет зависеть от концентрации реагентов, а только лишь от развитости поверхности взаимодействующих компонентов. Такие реакции имеют нулевой порядок по всем реагентам и для них υ0=υ0–k·τ , то есть, скорость реакции линейно уменьшается во времени.
С другой стороны, в гетерогенных процессах с участием жидких и твердых реагентов на их скорость оказывает влияние интенсивность доставки реагентов в реакционное пространство. Именно поэтому в выражении для скорости используется не объемная концентрация вещества Сμ(раствора), а молярная концентрация на поверхности твердой фазы в зоне реакции Сμ(S), ее принято еще обозначать символом Сs. С учетом сказанного, уравнение скорости гетерогенной реакции первого порядка примет вид:
υІ = kІ·Cs·S .
Перемешивание увеличивает скорость доставки реагентов и выравнивает концентрацию на поверхности реакционной зоны, поэтому гетерогенные процессы проводят при постоянном перемешивании.
Влияние температуры на реакционный процесс также оказывается достаточно сложным. Повышение температуры ускоряет большинство химических реакций, т.к. при этом увеличивается кинетическая энергия частиц, участвующих во взаимодействии, и возрастает скорость их перемещения в реакционном пространстве.
Согласно эмпирическому правилу Вант-Гоффа (1878г):
повышение температуры на каждые 100 увеличивает скорость химической реакции в 2÷4 раза.
Уравнение Вант-Гоффа имеет вид:
( Т2-Т1)/ 10
υT2/υT1 = γ . В этом уравнении υT2 и υT1 – скорости реакции при температурах Т2 и Т1, γ – температурный коэффициент Вант-Гоффа, принимающий, по определению, значения в пределах γ = 2÷4 .
Поскольку скорости и константы скоростей химической реакции пропорциональны друг другу, а время протекания реакции обратно пропорционально ее скорости, то уравнение Вант-Гоффа относительно констант и времени протекания реакции при различных температурах можно записать следующим образом:
Т2-Т1 Это уравнение можно использовать лишь для 10 расчетов в узком интервале температур, близких
kT2 ‗ τ1 ‗ γ к 100 0 С. Установлено, что температурный
kT1 τ2 коэффициент Вант - Гоффа для эндотермических
реакций выше, чем для экзотермических : γэнд>γэкз .
Более точным уравнением, показывающим взаимосвязь между константой скорости химического процесса и температурой, и действующим в неограниченном диапазоне температур, является уравнение С. Аррениуса (1889г, Швеция): k = A·e-Ea/RT. В этом уравнении: k – константа скорости химической реакции при температуре Т; А – стерический множитель, показывающий долю наиболее эффективных (приводящих к взаимодействию) столкновений между реагентами; Еа – энергия активации реагентов, зависящая от их природы (это та избыточная энергия, которой должны обладать частицы реагирующих веществ, чтобы между ними произошло взаимодействие). Для большинства химических реакций энергия активации Еа находится в пределах Еа = 40 ÷ 400 кДж/моль.
Как и все предыдущие кинетические уравнения, уравнение Аррениуса можно преобразовать в логарифмическую форму. Тогда оно приобретет вид:
ℓnk = ℓnA–Ea/RT. Как следует из уравнения, в координатах ℓnk ÷ 1/Т
график зависимости константы скорости от температуры представляет собой прямую линию, отсекающую на ординате отрезок, равный lnA, а тангенс угла наклона прямой к оси абсцисс позволяет определить энергию активации: tgα=–Ea/R (см. рис. 55).

 ℓnk
ℓnk




 ℓnA
ℓnA



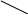

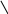


 1/T
1/T
Рис.55. Зависимость константы скорости реакции от температуры
Это уравнение используется при расчете постоянных для заданных химических реагентов величин А и Еа, и для мониторинга зависимости скорости химической реакции от температуры.