

Раздел III. ХИМИЧЕСКИЕ СИСТЕМЫ
Тема 9. ТЕОРИЯ РАСТВОРОВ
§1. Общая характеристика растворов, их классификация. Теория процесса растворения. Способы выражения состава раствора. Общие свойства растворов
Раствором называется многокомпонентная однофазная устойчивая система переменного состава.
Растворителем называют один из компонентов раствора, который при его образовании не меняет своего фазового состояния и присутствует, как правило, в наибольшем количестве.
В зависимости от их агрегатного состояния растворы могут быть твердыми (т), жидкими (ж), газообразными или газовыми (г). Все растворы относят к дисперсным системам, т.е. таким, в которых компоненты находятся в раздробленном состоянии.
Раствор является гомогенным, если частицы растворенного вещества не сохраняются в нем в виде отдельной фазы, имеющей реальную границу раздела. Если при этом размеры частиц не превышают ø1нм, то раствор считается истинным.
При увеличении размеров частиц в пределах от 1 до 100 нм раствор приобретает специфические свойства: на поверхности частиц появляется заряд за счет адсорбции из раствора определенных ионов. Такой раствор называют коллоидным. Свойствами коллоидных растворов обладают и растворы полимеров: они раздроблены до молекулярного состояния, но из-за больших размеров самих молекул ведут себя как коллоиды.
Гетерогенные системы, содержащие частицы размером ø >100 нм, являются грубодисперсными и кинетически неустойчивыми. К ним относятся пены (ж в г), эмульсии (ж в ж), суспензии (т в ж).
В настоящей теме мы не будем рассматривать газовые растворы (их свойства ничем не отличаются от свойств смеси газов) и твердые растворы (о поведении твердых растворов металлов – сплавах будет сказано в другом разделе). Наибольший и теоретический и практический интерес представляют жидкие растворы, на них мы и остановимся.
Используется несколько принципов классификации жидких растворов:
а) по исходному фазовому состоянию растворяемого вещества жидкие растворы бывают типов:
−(г в ж), например, раствор диоксида углерода СО2 в воде или раствор хлора Сl2 в воде;
−(ж в ж), например, растворы эфиров в спиртах, раствор серной кислоты Н2SO4 в воде;
− (т в ж), например, водные растворы солей, щелочей, твердых кислот, амальгамы металлов (растворы металлов в жидкой ртути Hg);
б) в зависимости от природы растворителя растворы классифицируют как протонные и апротонные:
- протонные растворы образованы растворителями, способными в силу своей полярности отщеплять протоны Н+, такие растворители называют протонными (к ним относятся вода Н2О, цианистая кислота НСN, уксусная кислота СН3СООН и др.). В протонных растворителях хорошо растворяются протофильные вещества, т.е. способные поглощать протоны Н+, это соли МеАn, основания Ме(ОН)n, основные оксиды МехОy;
- апротонные растворы образованы неполярными растворителями (к ним относятся бензол С6Н6, ацетон (СН3)2СО, хлороформ СН3Сl, диметилсульфоксид (СН3)2SО, диоксан С4Н8О2 и т.п.). В них наилучшим образом растворяются органические вещества, сами также являющиеся неполярными;
в) по величине диэлектрической проницаемости растворителя (ε) последние делятся на:
- полярные (высокая диэлектрическая проницаемость ε у воды Н2О, аммиака NН3∙Н2О, диметилфорамида (СН3)2NСНО и др.), в них растворяемые вещества обязательно диссоциируют на ионы;
- неполярные (низкая диэлектрическая проницаемость ε у четыреххлористого углерода СCl4, бензинов, керосинов и т.п. растворителей).
Самый распространенный и природный, и лабораторный, и промышленный растворитель – это вода Н2О, она полярная, протонная, с высокой диэлектрической проницаемостью ε, доступная и экономически выгодная. Однако вода плохо поддается полной очистке после удаления растворенных в ней веществ и загрязняет природную среду. Поэтому в последнее время формируется тенденция перехода на другие растворители, в основном органические, очистка и возвращение в производство которых (рекуперация) требуют меньших затрат, чем при очистке воды. Тем не менее, вода еще долго будет служить образцом надежного растворителя, активного с химической и удобного с потребительской точки зрения.
На процесс растворения, кроме природы растворителя, существенное влияние оказывают и другие факторы – агрегатное состояние растворяемого вещества, температура раствора и т.п.
Термодинамика процесса растворения
Согласно законам термодинамики процесс растворения вещества в жидкости может быть самопроизвольным только в том случае, если при этом уменьшается свободная энергия Гиббса (∆G) образующейся системы – раствора. Вспомним, что ∆G=∆H−T∆S и эта величина будет меньше нуля (∆G<0) при условии, что |∆H| < |T∆S|.
При растворении в жидкости твердых веществ их структура меняется, степень упорядоченности резко падает, а значит, энтропия такой системы возрастает (∆S>0). Вклад энтропийного фактора в свободную энергию будет тем больше, чем выше температура процесса, а, следовательно, с ростом температуры растворимость твердых веществ в жидкостях обязательно будет увеличиваться.
Наоборот, растворение в жидкости газообразного вещества приводит его структуру в более упорядоченное состояние (уменьшаются расстояния между молекулами, усиливаются межмолекулярные взаимодействия) и энтропия системы понижается (∆S<0). В этом случае, вклад энтропийного фактора в величину свободной энергии будет минимальным при низких температурах. Из этого следует, что растворимость газов в жидкостях увеличивается при охлаждении и уменьшается с ростом температуры.
Тепловой эффект (энтальпийный фактор) процесса растворения (∆Hраств) также принципиально важен. По своей сути, он является разницей между энергией, затрачиваемой на разрушение структуры растворяемого вещества (Естр), и энергией, выделяющейся в результате сольватации этих молекул молекулами растворителя (Есольв). Таким образом,
∆Hраств = Естр – Есольв .
При растворении газов тепловой эффект ∆Hраств = −Eсольв<0, т.к. у газов нет структурируемой фазы и для них Естр = 0. Отсюда следует, что растворение газов всегда является экзотермическим процессом.
У молекулярных кристаллов и жидкостей в структуре присутствуют только Ван-дер-ваальсовые взаимодействия, поэтому для них Естр = min и |Естр| < |Eсольв| . В силу указанных причин растворение, например, сахара, глицерина, спирта является также экзотермическим процессом.
В неполярных жидкостях неполярные молекулы не сольватируются (Есольв→0), у них и Естр также очень мала (Естр→0), т.к. нет взаимодействия между молекулами. В этом случае ∆H=0, т.е. растворение неполярных веществ в неполярных растворителях не сопровождается тепловым эффектом (например, при растворении йода J2 в четыреххлористом углероде CCl4) и скорость растворения будет определяться только энтропийным фактором.
При растворении ионных кристаллов происходит разрыв химических связей под действием молекул растворителя, в этом случае Естр>>0 и Естр>Есольв, а это значит, что ∆H>0 и процесс растворения является эндотермическим.
Поскольку процесс растворения является самопроизвольным, то он и обратимый, схематически это можно представить как
Растворение ◄───►Выделение растворенного вещества из раствора.
Процесс, обратный растворению твердых веществ, называют кристаллизацией, жидких – испарением, газовых – сублимацией. При выравнивании скоростей растворения и выделения фазы растворяемого вещества концентрация раствора становится постоянной при заданных внешних параметрах (Т=Const и P=Const). Такой раствор называют насыщенным, для него ∆С=0.
Известны вещества, которые при нагревании увеличивают свою концентрацию в растворе, но при очень медленном охлаждении не кристаллизуются. Такие растворы называют пересыщенными, они термодинамически неустойчивы и легко разрушаются при попадании в систему «затравки» - частичек пыли, стекла, кристалла соли или при механическом воздействии (встряхивании, перемешивании). Пересыщенные растворы были впервые получены в ХVIII веке Т. Ловицем и широко применялись для выращивания больших кристаллов.
Способы выражения состава раствора определяются степенью его насыщенности или концентрацией.
Концентрация насыщенного раствора является мерой растворимости (s) вещества при заданных внешних параметрах. Растворимость на практике выражают либо в массовой доле растворенного вещества по отношению к общей массе раствора (ω=m/m+m0, где m− масса растворенного вещества, m0−масса растворителя), либо в массе растворенного вещества, содержащегося в 100г растворителя (s=m/m0 при m0=100г).
По способности к растворению вещества делят на хорошо растворимые и мало растворимые. Не существует в природе абсолютно нерастворимых веществ.
Для измерения растворимости газов используют не их массу, а парциальное давление. По правилу У. Генри (1803), растворимость газа (sг) прямо пропорциональна его парциальному давлению: sг = kP̃, где sг – растворимость газа в жидкости; k – коэффициент пропорциональности, зависящий от природы газа и растворителя, а также от температуры растворения; Р̃− парциальное давление растворенного газа.
Концентрация растворов является их основной количественной характеристикой. Она отражает содержание растворенного вещества (или веществ, если их несколько) в единице массы или объема раствора (реже растворителя).
Наиболее часто употребляются на практике следующие способы выражения концентрации растворов:
а) молярная концентрация (обозначается Сμ или М) – показывает количество растворенного вещества (ν), содержащееся в единице объема раствора(V). Формула расчета: Сμ=ν/V (моль/л). Поскольку ν=m/M (отношение физической массы растворенного вещества m к его молярной массе М), то Сμ=m/M·V ;
б) молярная концентрация эквивалента (обозначается СN или Н) – показывает количество эквивалентов растворенного вещества (νэ), содержащееся в единице объема раствора (V). Формула расчета:
СN = νэ/V (моль/л). С учетом понятия эквивалента (νэ=m/Mэ, а также Mэ=M/B, где Мэ – молярная масса эквивалента растворенного вещества, М – его молярная масса, В – валентность в конкретном химическом соединении), получим еще одно выражение для расчета молярной концентрации эквивалента: СN = m·B/M·V (моль/л);
в) моляльная концентрация (обозначается Сm или м) – показывает количество растворенного вещества (ν), содержащееся в единице массы растворителя (m0). Формула для расчета: Сm=ν/m0 (моль/кг), или с учетом предыдущих подстановок, Сm=m/M·m0 (моль/кг);
г) молярная доля растворенного вещества (обозначается χ - кситта) – показывает отношение количества растворенного вещества (ν) к количеству вещества раствора (ν + ν0), где ν0− количество вещества растворителя. Формула для расчета: χ=ν/ν+ν0 .Молярная доля является безразмерной величиной и выражается в процентах, тогда окончательно χ=ν·100%/ν+ν0;
д) массовая доля растворенного вещества (обозначается ω - омега) – показывает отношение физической массы растворенного вещества (m) к массе всего раствора (m+m0). Формула для расчета: ω=m/m+m0. Так же как и молярная доля массовая выражается в процентах ω=m·100%/m+m0;
е) объемная доля растворенного вещества (обозначается φ – фи) – показывает отношение объема растворенного газа или жидкости (V) к объему всего раствора (V + V0). Формула для расчета: φ=V/V+V0 или
φ=V·100%/V+V0 ;
ж) титр (обозначается Т) – показывает отношение физической массы растворенного вещества (m) к единице объема раствора (V). Формула для расчета: Т = m/V (г/мл). Поскольку этот способ выражения концентрации применяется к очень разбавленным растворам и для ограниченного объема жидкостей, используемых в анализе, то объем измеряется в мл.
Общими свойствами растворов (коллигативными) называют такие их свойства, которые не зависят от природы растворяемого вещества и растворителя, а определяются только их концентрацией. В наибольшей степени они проявляются в разбавленных растворах нелетучих веществ. Эти растворы именуются идеальными, т.к. в них отсутствуют какие–либо взаимодействия между всеми молекулами (и растворяемого вещества, и растворителя).
Коллигативные свойства подчиняются определенным законам.
Законы Р. Рауля (1883-1885гг) рассматривают влияние молекул растворенного нелетучего вещества на давление молекул насыщенного пара растворителя над поверхностью раствора, по отношению к давлению молекул насыщенного пара над поверхностью чистого растворителя.
Понятно, что частицы растворенного вещества, скапливаясь у границы раздела фаз (ж) | (г), препятствуют испарению молекул жидкости, и, следовательно, понижают ее давление над поверхностью раствора.
I-й закон Рауля (тонометрический) гласит:
Понижение давления насыщенного пара растворителя над раствором пропорционально молярной доле растворенного вещества.
Математическое выражение I-го закона Рауля: Р0–Р = Р0·χ , где Р0 – давление насыщенного пара растворителя над чистым растворителем, Р – давление насыщенного пара растворителя над раствором, χ- молярная доля растворенного вещества. Если обозначить (Р0–Р) через ΔР, получим:
Р0–Р/Р0 = ΔР/Р0. Это отношение называют относительным понижением давления насыщенного пара растворителя над раствором. Закон будет выглядеть следующим образом: ΔР/Р0 = χ , и звучать несколько иначе:
Относительное понижение давления насыщенного пара растворителя над раствором равно молярной доле растворенного вещества.
II-й закон Рауля (эбуллиоскопический и криоскопический) гласит:
Повышение температуры кипения и понижение температуры кристаллизации раствора прямо пропорционально моляльной концентрации растворенного вещества.
Обозначим температуру кипения раствора через Ткип, температуру кипения растворителя через Токип, тогда повышение температуры кипения будет равно ΔТкип = Ткип – Токип. Аналогично, Ткр – температура кристаллизации раствора, Токр – температура кристаллизации растворителя и тогда понижение температуры кристаллизации ΔТкр = Токр – Ткр (изменение температуры должно оставаться положительной величиной). После введения указанных обозначений запишем математическое выражение II-го закона Рауля: ΔТкип=kэСm и ΔТкр=kкСm. В приведенных уравнениях Сm – моляльная концентрация раствора, kэ и kк – коэффициенты пропорциональности, имеющие специфические названия: kэ – эбуллиоскопическая постоянная (от греч. еbulios – жар), kк – криоскопическая постоянная (от греч. сrios – холод). Физический смысл этих коэффициентов – они показывают изменение температуры кипения и кристаллизации раствора при концентрации, равной 1моль/л.
 Теоретически
объяснить законы Рауля можно с помощью
анализа диаграммы состояния растворителя
- воды и раствора (см. рис.60).
Теоретически
объяснить законы Рауля можно с помощью
анализа диаграммы состояния растворителя
- воды и раствора (см. рис.60).
И з
физики известно, что жидкость закипает
в момент, когда давление ее паров внутри
капель жидкости становится равным
атмосферному давлению. Поскольку
давление насыщенного пара над раствором
меньше, чем над чистым растворителем
(кривая 2 на рис.60), то для достижения
состояния равенства давлений необходима
дополнительная температура. Аналогично,
жидкость кристаллизуется в твердую
фазу, когда давление насыщенного пара
над ней уравнивается с давлением паров
над твердой фазой. Как видно на рис.60,
такое состояние для раствора наступает
при более низкой температуре, чем для
чистого растворителя.
з
физики известно, что жидкость закипает
в момент, когда давление ее паров внутри
капель жидкости становится равным
атмосферному давлению. Поскольку
давление насыщенного пара над раствором
меньше, чем над чистым растворителем
(кривая 2 на рис.60), то для достижения
состояния равенства давлений необходима
дополнительная температура. Аналогично,
жидкость кристаллизуется в твердую
фазу, когда давление насыщенного пара
над ней уравнивается с давлением паров
над твердой фазой. Как видно на рис.60,
такое состояние для раствора наступает
при более низкой температуре, чем для
чистого растворителя.
Законы Рауля широко используются для практического определения молярной массы неизвестных веществ и их идентификации. Преобразование II-го закона, например, дает расчетную формулу: k·m·103
М = ——— .
ΔT·m0
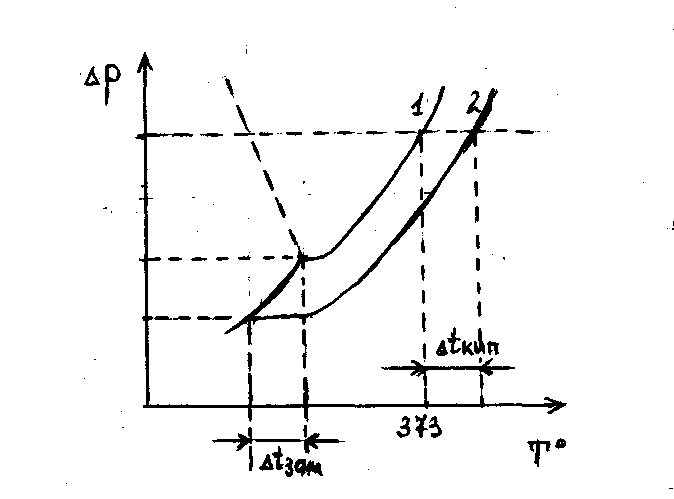
Рис. 60. Диаграмма состояния насыщенного пара растворителя (кривая 1)
и раствора (кривая 2).
Наличие осмотического давления является еще одним коллигативным свойством разбавленных растворов.
Самопроизвольный переход растворителя через полупроницаемую мембрану, разделяющую раствор и растворитель или два раствора с разной концентрацией, называется осмосом.
Осмос обусловлен диффузией, т.е. энтропийным фактором выравнивания концентрации по всему объему системы. Но поскольку поры в мембране очень маленькие и способны пропускать через себя только небольшие молекулы растворителя и задерживать молекулы растворенного вещества, то осмос всегда направлен от растворителя к раствору или в раствор с большей концентрацией из раствора с меньшей концентрацией.
Сила, под действием которой осуществляется осмос, называется осмотическим давлением. Ясно, что осмотическое давление будет тем выше, чем больше разница концентраций растворов по обе стороны мембраны. Осмос является самопроизвольным процессом, и он продолжается до тех пор, пока давление, под действием которого частицы растворителя проникают через мембранную перегородку, не уравновесится с атмосферным давлением. Можно искусственно создать такие условия, чтобы внешнее давление значительно превышало осмотическое. Тогда в системе можно вызвать «обратный осмос»: молекулы растворителя будут переходить через мембрану из более концентрированного раствора в менее концентрированный. Обратный осмос энергетически невыгоден, требует больших затрат дополнительной энергии, но используется при очистке сточных вод, при получении питьевой воды из морской в критических условиях и т.п.
В 1887 г. Я. Вант-Гофф установил закон осмотического давления:
Осмотическое давление раствора прямо пропорционально его молярной концентрации и температуре.
Математическое выражение закона Вант-Гоффа: π = СµRT, здесь π–осмотическое давление; Сµ–молярная концентрация раствора; Т – температура, R – коэффициент пропорциональности, равный универсальной газовой постоянной Ридберга (R= 8,31 Па.м3/моль.К).
Осмос имеет важное биологическое значение, т.к. обеспечивает доступ растворителя (воды) в клетки растений и животных. Растворы с одинаковым осмотическим давлением изотоничны, если осмотическое давление внутри клеток выше атмосферного, то оно является гипертоническим, если ниже – то гипотоническим. Например, при повышенной температуре тела человека t=380С осмотическое давление в крови составляет 780кПа, что значительно выше атмосферного и человек болеет.
Законы Рауля и Вант-Гоффа выведены эмпирическим путем и действуют в узком интервале концентраций до Сµ≤0,01 моль/л. При более высоких концентрациях и при растворении в воде молекул полярного строения законы Рауля и Вант-Гоффа для таких растворов не выполняются.
Отклонение большинства истинных растворов от законов Рауля и Вант-Гоффа обусловлено взаимодействием между частицами растворенного вещества, а также между частицами растворенного вещества и растворителя. Учет этого взаимодействия не всегда можно реально оценить из-за его чрезвычайной сложности.
Позднее, для приведения законов Рауля и Вант-Гоффа в соответствие с поведением истинных растворов было решено использовать для всех типов растворов понятие активности вместо понятия концентрации.
Активность – это величина, показывающая во сколько раз действующая концентрация раствора меньше его истинной концентрации. Активность связана с концентрацией соотношением: а=γСµ, где γ–коэффициент активности раствора, а Сµ– его молярная концентрация. Коэффициент активности вычисляется из экспериментальных данных:
γ = ΔTкр(практ)/ΔTкр(теор) = ΔTкип(практ)/ΔTкип(теор) ≤1.
Физический смысл активности будет понятен несколько позже.
§2. Химическое равновесие в растворах: сольватация, диссоциация, ионные равновесия. Сильные и слабые электролиты
В реальных растворах между молекулами растворителя и частицами растворяемого вещества всегда присутствует довольно значительное взаимодействие, влияющее на свойства раствора. Это взаимодействие называют сольватацией, в случае водного растворителя – гидратацией.
Процесс сольватации (гидратации) включает в себя три последовательно-параллельные стадии:
I-я стадия – молекулярная диссоциация. Под действием Ван-дер-ваальсовых сил, направленных от растворителя к растворяемому веществу, последнее разделяется до молекулярного уровня, распространяется по объему растворителя и обволакивается его молекулами, образуя сольватированные (гидратированные) молекулы растворяемой фазы – сольваты (гидраты). На схеме и рис.61 представлен этот процесс:
[AnBm]Х + Y( s♂) ──► X{[AnBm]•Y(s♂)}
Кристалл из X Y молекул Х молекул соли, каждая
молекул соли растворителя из которых окружена Y
АnBm s♂ молекулами растворителя

Рис. 61. Структура молекулярного сольвата.
II-я стадия – электролитическая диссоциация. Процесс возможен только в полярных растворителях, под действием которых в молекулах растворяемого вещества происходит разрыв химических связей (энтальпийный фактор) с образованием положительных и отрицательных ионов, каждый из которых также покрывается сольватной оболочкой (см. рис.62):
[AmBn]•у(s♂) ──► [An+]•n(s♂) + [Bm¯]•m(s♂)
Сольват молекулярный сольват катиона сольват аниона
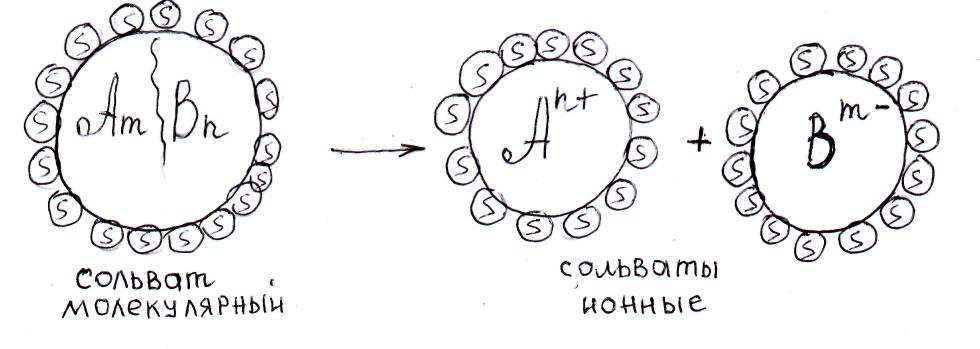
сольватированных ионов.
III-я стадия – диффузия сольватированных агрегатов (сольватов) по всему объему раствора (энтропийный фактор).
В зависимости от внешних условий, природы растворяемого вещества и растворителя, сольватация может быть полной, либо с исключением любой из указанных стадий.
Например, при растворении в воде неполярных молекул сахара или кислорода, в четыреххлористом углероде – этилена и т.п., процесс сольватации не будет в себя включать стадию II. В результате полученный раствор не будет обладать свойствами электропроводности, при небольших концентрациях в нем будут выполняться законы Рауля и Вант-Гоффа. Такой раствор называют неэлектролитом.
При полной сольватации система будет обладать электрической проводимостью, ее называют электролитом.
Явление разложения молекул вещества на ионы под действием молекул растворителя (электролитическая диссоциация) впервые было изучено С. Аррениусом в 1887г и известно в науке как теория электролитической диссоциации Аррениуса.
Основные положения этой теории заключаются в следующем.
1). Процесс разложения вещества на ионы разного заряда является обратимым,
AmBn + (p+q)s♂ ◄═══►m[An+]·p(s♂) + n[Bm-]·q(s♂) .
Следует заметить, что коэффициенты р и q перед молекулами растворителя (s♂) меняются с изменением концентрации раствора, температуры и других параметров, в том числе – природы растворяемых веществ.
2). Если AmBn по своей природе является ионным соединением, то процесс диссоциации становится необратимым:
AmBn + (p+q)s♂ ═══ m[An+]·p(s♂) + n[Bm-]·q(s♂).
В этом случае в уравнениях диссоциации, как правило, опускают сольватант (s♂) и его запись упрощается. Например, уравнения диссоциации щелочей и солей в общем виде можно записать следующим образом:
Me(OH)n ═ Men+ + nOH–
MenAnm ═ nMem+ + mAnn–.
Растворы, содержащие растворенные вещества в полностью ионизированном виде, называют ионными проводниками II рода (проводниками I рода, как мы знаем, являются металлы).
3). Если в процессе сольватации диссоциирует на ионы только часть молекул растворенного вещества, то их доля (ее называют степенью электролитической диссоциации и обозначают греческой буквой α - альфа) указывает на силу электролита. Из самого понятия степени диссоциации следует, что α=N/N0≤1, где N – количество диссоциированных на ионы молекул растворенного вещества, N0–общее число растворенных молекул. Электролит называют сильным, если степень его диссоциации α =1 и слабым, если α <1. Однако четкой границы при классификации силы электролитов быть не может, т.к. при замене растворителя степень диссоциации меняется значительно.
4). Диссоциация слабых электролитов является обратимым процессом, а поэтому к нему можно применить закон действующих масс для определения константы химического равновесия (называемой в данном случае константой электролитической диссоциации kp=kd). Чтобы не вносить дополнительные сложности, в записи уравнения электролитической диссоциации опустим сольватную оболочку из молекул растворителя (s♂). Тогда при растворении любого вещества с ковалентной полярной связью в полярном растворителе процесс его диссоциации подчиняется уравнению
AmBn <=> mAn+ + nBm- и по закону действующих масс
[An+]m·[Bm־]n
kp = kd = ——————— .
[AmBn]
Чем меньше будет значение константы диссоциации, тем прочнее молекулы растворяемого вещества и тем слабее образуемый при их растворении электролит.
Для определения зависимости степени диссоциации α от константы диссоциации kd упростим условную запись молекулы вещества до АВ и обозначим молярную концентрацию раствора этого вещества как Сµ. Тогда уравнение диссоциации будет выглядеть АВ<=>A+ + B־, выражение для константы диссоциации [A+]·[B־]
kd = —————. С учетом введенных обозначений α
[AB]
и Сµ, равновесные концентрации ионов и недиссоциированных молекул будут равны: α=[A+]/Cµ, следовательно, [A+]=α·Cµ. Из уравнения реакции видно, что [A+]=[B־]=αCµ, а [AB]=Cµ– αCµ = Cµ(1–α).
Подставив полученные выражения в уравнение для константы диссоциации, получим в окончательном виде:
α2Cµ
kd = ———.
1 – α
Приведенное уравнение зависимости константы диссоциации электролита от степени диссоциации и концентрации раствора широко известно в практике как закон разведения К. Оствальда (1888):
С увеличением концентрации электролита степень его диссоциации всегда уменьшается и наоборот, степень диссоциации электролита тем больше, чем он более разбавлен.
Теория поведения сильных электролитов была разработана гораздо позднее теории электролитической диссоциации С. Аррениуса, ее авторами стали норвежские ученые П. Дебай и Э. Хюккель (1927).
В растворах сильных электролитов диссоциация практически полная и этот процесс необратим. Уравнения диссоциации, как было отмечено выше, имеют вид: NaOH = Na+ + OH־ ;
Al2(SO4)3 = 2Al3+ + 3SO42־.
Накопление заряженных частиц в растворе повышает его электрическую проводимость. Несмотря на то, что каждый ион окружен сольватной оболочкой из молекул растворителя, силы электростатического взаимодействия между ионами противоположного знака заставляют их координироваться друг относительно друга. В результате вокруг каждого иона возникает своеобразная ионная атмосфера, состоящая из молекул растворителя и сольватированных ионов противоположного знака.
На рис. 63 представлена ионная атмосфера, образовавшаяся вокруг центрального положительно заряженного иона из молекул растворителя и отрицательных ионов противоположного знака, окруженных, в свою очередь, и молекулами растворителя, и положительными ионами.
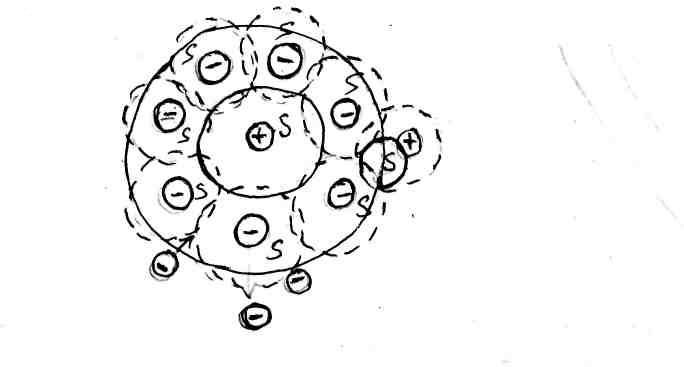









Рис. 63. Схема строения ионной атмосферы: 1 – центральный ион А+;
2 – сольватная оболочка из молекул растворителя; 3 – ионы В־;
4 – сольватная оболочка из молекул растворителя вокруг отрицательных ионов.
Величина заряда ионной атмосферы равна заряду центрального иона, но противоположна ему по знаку. В результате теплового движения частиц ионная атмосфера крайне подвижна, в ней постоянно происходит обмен ионами и молекулами растворителя. При этом все основные параметры раствора выражаются в виде функции суммарного взаимодействия всех входящих в его состав ионов с их ионными атмосферами.
С увеличением концентрации раствора сильного электролита плотность заряда ионной атмосферы возрастает, а ее средний радиус уменьшается, что еще больше повышает энергию взаимодействия противоположных по знаку ионов. Вместо диффундирующих по всему объему ионных атмосфер, возникают ионные ассоциаты:
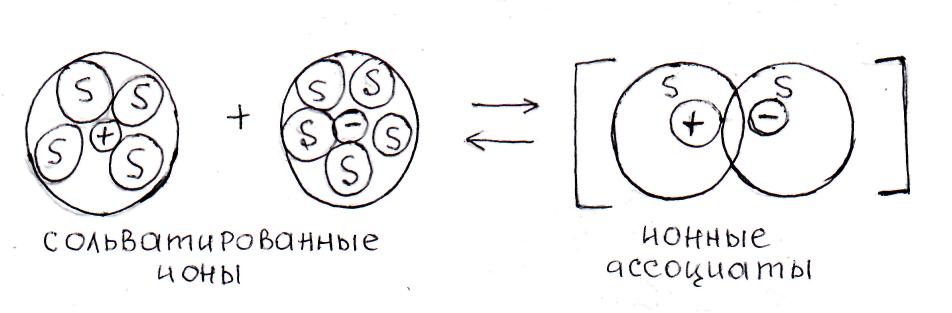
В отличие от недиссоциированной молекулы, в которой частицы соединены силами ковалентной полярной или ионной химической связи, в ионных ассоциатах присутствуют только силы электростатического взаимодействия между ионными атмосферами. Тем не менее, появление ионных ассоциатов вызывает снижение электрической проводимости растворов вследствие возрастания его сопротивления прохождению электрического тока.
При дальнейшем повышении концентрации образуются ионные тройники, ионные квадруполи, что еще больше снижает электропроводность раствора. Например, в концентрированном водном растворе хлорида калия KCl присутствуют ионные ассоциаты, состоящие из: ионных тройников (KCl2־ и K2Cl+); ионных квадруполей (K2Cl 2; KCl32־; K3Cl2+) и т.п.
Таким образом, свойства растворов сильных электролитов зависят не только от природы растворяемого вещества и растворителя, но и от концентрации раствора. Совокупность поведения всех типов ионных ассоциатов выражают ионной силой раствора (J), определенной Дебаем и Хюккелем как полу сумма произведений концентрации всех ионов, присутствующих в растворе, на квадрат их заряда:
Сµ(А+)·Z2(A+) + Cµ(В־)·Z2(B־) + … Cµ(i)·Z2(i)
J = ————————————————— = —————.
2 2
Правило ионной силы: ионы одинаковой зарядности, независимо от их природы, в разбавленных растворах с одинаковой ионной силой имеют равные коэффициенты активности.
При этом предполагается, что среднее значение коэффициента активности электролита (γ±) определяется как среднее геометрическое коэффициентов активности образующих его ионов. Например, для сильного электролита AmBn, диссоциирующего на ионы по схеме
AmBn ═ mAn+ + nBm- ,
усредненный коэффициент активности (γ±) будет определяться по формуле:
____________
γ±=(m+n)√ γm(An+)·γn(Bm-).
Проиллюстрируем применение этой формулы к уравнениям электролитической диссоциации солей KCl и Al2(SO4)3:
_________ ______________
для KCl γ±=√γ(K+)·γ(Cl-) ; для Al2(SO4)3 γ±=5√γ2(Al3+)·γ3(SO42-).
Ионные равновесия.
При составлении уравнений электролитической диссоциации сильных и слабых электролитов необходимо усвоить несколько правил, которые позволяют решить любую практическую задачу.
Правило I. В силу необратимости процессов диссоциации сильных электролитов, независимо от состава их молекул, уравнение диссоциации записывается только в одну ступень, без указания сольватной оболочки.
Например, диссоциация щелочей NaOH = Na+ + OH–,
RbOH = Rb+ + OH-;
Ва(ОН)2=Ва2+ + 2ОН-;
Са(ОН)2=Са2+ + 2ОН-;
Sr(OH)2=Sr2+ + 2OH-;
диссоциация сильных кислот HCl = H+ + Cl–,
HJO4 = H+ + JO4–;
диссоциация солей Со(NO3)2=Co2+ + 2NO3-;
Fe2(SO4)3 = 2Fe3+ + 3SO42-,
в том числе и комплексных солей [Cu(NH3)4](NO3)2 = [Cu(NH3)4]2+ + 2NO3–.
Правило II. Слабые электролиты всегда диссоциируют обратимо и ступенчато, каждой ступени диссоциации соответствует свое значение константы диссоциации (kd) и степени диссоциации (α). Приведем примеры ступенчатой диссоциации некоторых слабых электролитов с указанием ступенчатых констант диссоциации.
Диссоциация угольной кислоты по I-й ступени
Н2СО3 <=> Н+ + НСО3– , kd¹=4,5·10-7,
диссоциация по II-й ступени
НСО3– <=> Н+ + СО32-, kd²=4,8·10-11.
Диссоциация слабого основания гидроксида свинца (II) Pb(OH)2 по I-й ступени Pb(OH)2 <=> Pb(OH)+ + OH-, kd¹= 9,55·10-4;
диссоциация по II-й ступени Pb(OH)+ <=> Pb2+ + OH-, kd² = 3,0·10-8.
Как видим, каждая следующая ступень ослабляет электролит на несколько порядков, при этом среднее значение константы его диссоциации определяется как произведение констант ступенчатой диссоциации:
kd = kd¹·kd²·kd³· …
Правило III. Основания щелочноземельных металлов IIA группы Ве(ОН)2 и Mg(OH)2 и все многоосновные кислоты диссоциируют ступенчато, но по первой ступени диссоциации они - сильные электролиты, а по следующим – слабые.
Например, гидроксид магния Mg(ОН)2 по I-й ступени диссоциирует как сильный электролит Mg(ОН)2 = Mg(ОН)+ + ОН- ,
а по II-й ступени – как слабый Mg(ОН)+ <=> Mg2+ + ОН-.
Ортофосфорная кислота Н3РО4 является сильным электролитом при диссоциации по первой ступени Н3РО4 = Н+ + Н2РО4–, но слабым электролитом при диссоциации по второй и третьей ступени:
II-я ступень Н2РО4- <=> НРО42- + Н+,
III-я ступень НРО42- <=> РО43- + Н+.
Правило IV. Комплексные ионы также способны к диссоциации, но всегда – ступенчатой. Однако если при диссоциации комплексный ион отщепляет нейтральный лиганд, то константы диссоциации по ступеням почти не изменяются.
Например, ступенчатая диссоциация катиона диамминосеребра [Ag(NH3)2]+ будет выглядеть следующим образом:
I-я ступень [Ag(NH3)2]+ <=> [Ag(NH3)]+ + NH3, kd¹= 1,5·10-4;
II-я ступень [Ag(NH3)]+ <=> Ag+ + NH3 , kd² = 6,3·10-3.
При диссоциации комплексных ионов с высвобождением заряженных лигандов константы их диссоциации постепенно увеличиваются по мере уменьшения заряда комплексного иона.
Например, диссоциация комплексной соли хлорида гексагидроксожелеза(III) [Fe(OH)6]Cl3 по первой ступени будет являться необратимым процессом диссоциации сильного электролита
[Fe(OH)6]Cl3 ═ [Fe(OH)6]3+ + 3Cl- .
В дальнейшем диссоциации подвергается комплексный ион с выделением заряженного лиганда – иона ОН-. Процесс становится обратимым и ступенчатым, причем, отрыв каждого последующего иона ОН- становится все более легким: [Fe(OH)6]3+ <=> [Fe(OH)5]2+ + OH-,
[Fe(OH)5]2+ <=> [Fe(OH)4]+ + OH-,
[Fe(OH)4]+ <=> [Fe(OH)3]↓ + OH-.
Константы диссоциации комплексных ионов часто называют константами их нестойкости.
Ионный обмен в растворах электролитов
Поскольку в растворах полярных растворителей большинство растворенных веществ находятся в ионном состоянии, то и все реакции в растворах фактически являются обменными, если не изменяются степени окисления компонентов раствора. При этом возможны различные варианты обменных процессов.
Случай 1. В равновесных гомогенных системах не происходит химического взаимодействия по типу реакций обмена, в них имеет место только процесс сольватации всех компонентов раствора.
Например, при смешивании растворов хлорида натрия NaCl и нитрата калия KNO3 нельзя ожидать образования солей нитрата натрия NaNO3 и хлорида калия KCl по реакции NaCl + KNO3 = NaNO3 + KCl. Такая реакция невозможна (≠), т.к. при диссоциации солей реагентов в растворе будут присутствовать только их сольватированные ионы. Это легко доказать, если переписать уравнение в ионном виде (c учетом силы электролитов):
Na+ + Cl- + K+ + NO3- <=> Na+ + NO3- + K+ + Cl- .
Как видим обе части уравнения не изменились, значит, химическое взаимодействие отсутствует.
Случай 2. Если при смешивании электролитов образуется малорастворимый продукт (газ или твердая фаза), то такой ионный обмен становится необратимым и направлен в сторону уменьшения свободной энергии Гиббса (∆G<0).
Примеры таких реакций ионного обмена:
а) СаСl2 + Na2CO3 ═ CaCO3↓ + 2NaCl, или в сокращенном ионном виде
Са2+ + СО32- ═ СаСО3↓, ∆G° = -14,3 ккал/моль.
б) Na2CO3 + 2HCl ═ 2NaCl + H2O + CO2↑, или в сокращенном ионном виде СО32- + 2Н+ ═ СО2↑ + Н2О, ∆G° = -25,0 ккал/моль.
Случай 3. Процесс ионного обмена становится обратимым, если его продуктами являются ассоциированные или комплексные ионы (слабый электролит).
Примеры обратимых ионных реакций:
а) 2KCN + H2SO4 <=> 2HCN + K2SO4, или в сокращенном ионном виде CN- + H+ <=> HCN; ∆G° = -12,5 ккал/моль;
б) Hg(NO3)2 + 4KCN <=> K2[Hg(CN)4] + 2KNO3, или в ионном виде Hg2+ + 4CN- <=> [Hg(CN)4]2- ; ∆G°= -53,0 ккал/моль;
в) все реакции нейтрализации, т.к. они сводятся к сокращенному ионному уравнению Н+ + ОН- <=> Н2О ; ∆G°<0 .
Случай 4. Если в обеих частях уравнения ионного обмена есть малорастворимые или слабо диссоциирующие вещества, то процесс остается обратимым и равновесие сдвигается в сторону образования менее диссоциированного продукта (по-прежнему в сторону той реакции, при которой ∆G<0).
Примеры таких обратимых процессов запишем только в сокращенном ионном виде:
а) NH4OH + H+ <=> NH4+ + H2O, ∆G° = -20,0 ккал/моль;
б) [Ag(NH3)2]+ + 2H+ <=> Ag+ + 2NH4+, ∆G° = -12,5 ккал/моль;
в) СаСО3(т) + Н+ <=> Са2+ + НСО3- , ∆G° = -2,5 ккал/моль;
г) AgCl(т) +КJ(р-р) <=> AgJ(т) + КCl(р-р) или в ионном виде
AgCl + J- <=> AgJ↓+ Cl- , ∆G° = -12,7 ккал/моль.
Чтобы оценить, в какую сторону будет сдвигаться равновесие в подобных реакциях и правильно их записать, нужно предварительно рассчитать для них изменение энергии Гиббса при стандартных условиях.
§3. Частные случаи химического равновесия в растворах электролитов: электролитическая диссоциация воды, водородный показатель рН; теория действия индикаторов; современные теории кислот и оснований; произведение растворимости ограниченно растворимых соединений