

6 курс / Клинические и лабораторные анализы / Лабораторная_диагностика_туберкулеза_Ерохин_В_В_ред_
.pdfТема 8.2. «Глобальные филогенетические группы микобактерий туберкулезного комплекса»
Количество аудиторных часов – 1
Примерный план лекции
Основные вопросы, освещаемые в лекции:
•Представление о филогении микобактерий туберкулезного комплекса
•Три принципиальные генетические группы МБТ
•Основные линии МБТ
|
Вводная часть (5–10 мин) |
|
|
M.tuberculosis является членом микобактерий туберкулезного |
|
|
комплекса (МБТК), группы близко родственных видов (M.tuber- |
|
|
culosis, M.africanum, M.microti, M.bovis, M.canettii, M.pinnipedii, |
|
|
M. caprae и т.д.), которые могут вызывать туберкулез у человека и |
|
|
животных. |
|
|
Представители МБТК – высоко сходные родственные мико- |
|
|
бактерии, демонстрирующие значительный уровень гомологии на |
|
|
нуклеотидном уровне, несмотря на вариабельность в патогенно- |
|
|
сти, географическом распространении, некоторых физиологиче- |
|
|
ских свойствах, таких как морфология колоний, спектр чувстви- |
|
|
тельности к антимикробным препаратам, эпидемиология и спектр |
|
|
хозяев. Аллельный полиморфизм среди изолятов МБТК экстре- |
|
|
мально редок, встречается 1 на 10 000 п.о. |
|
|
Для установления отличий между микобактериями туберкулез- |
|
|
ного комплекса применяются сравнительные геномные исследова- |
|
|
ния. |
|
|
В одном случае сравнительные геномные исследования на- |
|
|
правлены на выявление однонуклеотидных полиморфизмов |
|
РЕКОМЕНДАЦИИ |
(ОНП), присущих данному виду или группе штаммов. Для исклю- |
|
чения влияния естественного отбора на распространение в по- |
||
|
||
|
пуляции какого-либо ОНП в качестве маркеров для межгеномного |
|
|
сравнения используют или функционально нейтральные нсОНП, |
|
|
или, чаще, сОНП, которые никогда не отражаются на жизнедея- |
|
|
тельности микроорганизма, т.к. никогда не приводят к изменению |
|
МЕТОДИЧЕСКИЕ |
аминокислотных последовательностей. |
|
В другом случае межгеномное сравнение направлено на вы- |
||
|
||
|
явление специфических делеций, характеризующих вид или штам- |
|
|
мовую группу. Учитывая, что геномные делеции рассматриваются |
|
|
как однонаправленные генетические события, сравнительный |
168
анализ делеций – это важный компонент исследования филогенетических взаимоотношений среди микобактерий МБТК.
Основная часть (30 мин)
Благодаря применению сравнительных геномных исследований стало возможным описать филогению штаммов M.tuberculosis и МБТК.
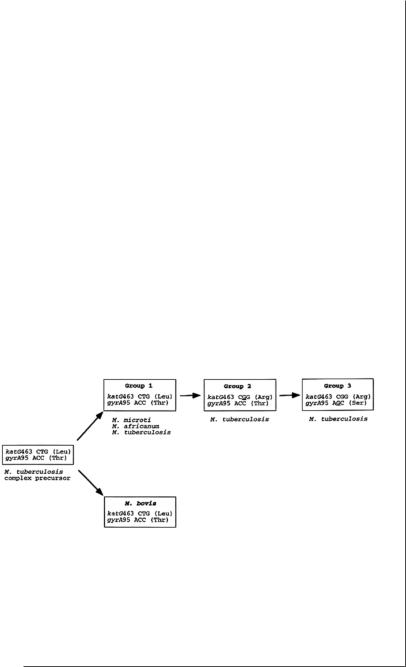
Принципиальные генетические группы (S. Sreevatsan). Для изучения эволюционного родства между штаммами M.tuberculosis S.Sreevatsan с соавторами использовали два функционально нейтральных нсОНП в кодоне 463 (Leu463Arg) гена katG, кодирующего каталазу-пероксидазу, и кодоне 95 гена gyrA (Thr95Ser), кодирующего субъединицу А ДНК-гиразы. На основе распределения полиморфизмов в этих двух локусах была создана трихотомическая система классификации, и современные микобактерии туберкулезного комплекса были разделены на три принципиальные генетические группы (ПГГ), названные 1, 2, и 3. ПГГ1 характеризовалась последовательностями katG463 CTC (Leu), gyrA95 ACC (Thr). Для ПГГ2 были характерны последовательности katG463 CGG (Arg), gyrA95 ACC (Thr). ПГГ3 имело последовательности katG463 CGG (Arg), gyrA95 AGC (Ser).
Такое распределение нсОНП по группам показывало, что ПГГ 1 является предковой для ПГГ 2, а ПГГ 2 – для ПГГ 3.
Рис. 1. Основные генетические группы микобактерий (Sreevatsan et al.,1997).
К первой принципиальной генетической группе были отнесены штаммы M.tuberculosis, M.bovis, M.microti, M.africanum, ко второй и третьей относились только штаммы M.tuberculosis.
Была получена высокая степень совпадения между группами, полученными типированием по ОНП, и молекулярно-эпидемио- логическими штаммовыми кластерами, установленными типиро-
РАЗДЕЛ 1. Лекционные материалы
169
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
ванием по ПДРФ IS6110, сполиготипированием и MIRU-VNTR, и штаммы со сходными профилями никогда не принадлежали к разным ПГГ. В то же время внутри одной ПГГ выявлялись штаммы, существенно отличавшиеся по генотипам, установленным традиционными молекулярно-эпидемиологическими методами типирования. Это указывает на то, что произошла ранняя дивергенция на ПГГ от общего предка в ходе эволюции вида M.tuberculosis, а варианты, выявляемые молекулярно-эпидемиоло- гическими методами типирования, возникли уже после дивергенции штаммов по этим трем ПГГ.
Учитывая, что распределение по этим основным генетическим группам базировалось только на двух нсОНП, были необходимы дополнительные исследования, дающие более подробную информацию о филогенетических связях современных штаммов M.tuberculosis.
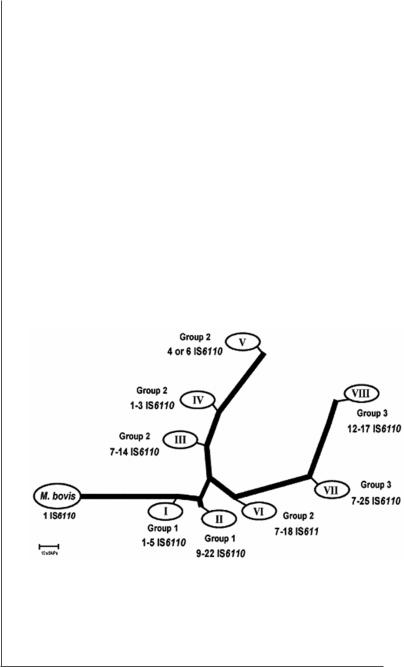
Основные линии МБТК на основе различий сОНП. Так как сОНП функционально нейтральны и просты для анализа, они являются подходящими маркерами для крупномасштабных популяционных молекулярно-генетических исследований, изучающих эволюционное родство среди бактериальных штаммов.
Рис. 2. Филогенетическое дерево построено по результатам анализа 148 сОНП с использованием метода объединения ближайших соседей (Gutacker et a.l, 2002). Показано 8 основных кластеров (I–VIII). Обозначения групп соответствуют 3 ПГГ M.tuberculosis (Sreevatsan et al., 1997). Цифрами обозначено число IS6110 элементов у штаммов, входящих в группу
170

M.M. Gutacker с соавторами с целью проведения детального филогенетического анализа изучили сОНП 432 штаммов МБТК (M.tuberculosis H37Rv, M.tuberculosis CDC1551, M.tuberculosis 210, и
M.bovis AF2122/97) по 148 сОНП. В результате было выделено восемь отдельных линий M.tuberculosis. Проведенный позднее филогенетический анализ 5069 клинических штаммов M.tuberculosis, полученных в результате 4 популяционных исследований с использованием 36 наиболее информативных сОНП, выявил дополнительную девятую линию (IIA).
При сопоставлении полученных результатов с принадлежностью к ПГГ было показано, что линии I и II относились к первой ПГГ, 2 группа была представлена III, IV, V, VI линиями, 3 группа – VII и VIII линиями (рис. 2). Другие представители МБТК, такие как M.bovis, M.microti, M.africanum, относящиеся к первой ПГГ, представляют отдельные группы, среди которых можно выделить 2 основные – это классический M.bovis и штаммы M.bovis, M.microti, M.africanum, занимающие промежуточное положение между группами штаммов M.tuberculosis и классическим M.bovis.
Эти исследования подтвердили наличие трех ПГГ, предложенных ранее S.Sreevatsan с соавторами. Другие исследователи также использовали сОНП анализ для изучения филогенетической структуры вида M.tuberculosis и получили результаты, хорошо согласующиеся с описанными выше.
Делеционный анализ. По результатам делеционного анализа, проведенного независимо рядом исследователей, было выявлено порядка двух десятков регионов различия (RD, regions of difference) у штаммов МБТК.
Например, при проведении сравнительных геномных исследований штаммов M.tuberculosis H37Rv и M.bovis BCG было выявлено 16 регионов, которые отсутствовали у M.bovis BCG (делеции RD116) по сравнению с M.tuberculosis H37Rv. Параллельно было описано 5 регионов, отсутствующих у H37Rv, по сравнению с M.bovis BCG (делеции RvD 1-5).
Кроме того, описана делеция, специфическая для "современных" M.tuberculosis TbD1. Она характеризуется отсутствием фрагмента размером 2153 п.о., затрагивающего гены mmpS6 и mmpL6.
Штаммы МБТК, имеющие делецию TbD1, рассматриваются как представители «современного» типа микобактерий, и могут относиться к любой ПГГ по классификации Sreevatsan (1997). Микобактерии, имеющие данный фрагмент в геноме, были представителями только первой ПГГ.
РАЗДЕЛ 1. Лекционные материалы
171
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
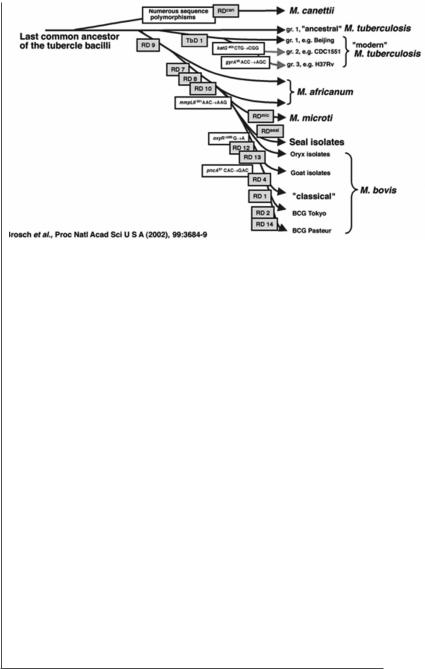
Рис. 3. Схема предполагаемого эволюционного пути микобактерий, демонстрирующая потерю ДНК у некоторых филогенетических линий (Brosch, Gordon et al., 2002).
На основе анализа делеций Brosch с соавторами была предложена схема предполагаемого эволюционного пути микобактерий, демонстрирующая потерю ДНК у некоторых филогенетических линий (рис. 3). Было показано, что штаммы M.tuberculosis и M.bovis являются хорошо дифференцированными друг от друга и расположены на обособленных ветвях филогенетического дерева. Между этими двумя видами расположены M.africanum, M.microti и M.pinnipedii. Штаммы M.canettii располагаются на обособленной ветви по отношению ко всем вышеперечисленным штаммам, что, очевидно, говорит о том, что произошла ранняя дивергенция штаммов M.canettii и гипотетического общего предка для M.tuberculosis, M.bovis, M.africanum, M.microti и M.pinnipedii от предка, давшего начало всем микобактериям туберкулезного комплекса. Полученные данные опровергли существующую до недавнего времени гипотезу, что штаммы M.bovis являются непосредственными предками M.tuberculosis, и показали, что M.bovis и M.tuberculosis представляют собой две обособленные ветви филогенетического дерева.
Что касается вида M.tuberculosis, то характерный для штаммов M.tuberculosis высокий уровень консервативности генов "домашнего хозяйства", малое количество синонимичных мутаций и данные геномного делеционного анализа позволили предположить,
172

что современные штаммы M.tuberculosis в процессе видообразования образовали свою ветвь приблизительно 15.000-20.000 лет назад. Недавняя работа M.C.Gutierrez пролила свет на эволюционный возраст M.tuberculosis. В этой работе исследователи классически секвенировали части шести генов "домашнего хозяйства" (katG, gyrA, gyrB, rpoB, hsp65 и sodA) и полный ген 16рРНК девяти штаммов M.tuberculosis и семи штаммов M.tuberculosis, дающих гладкие колонии. Последняя группа штаммов, выделенная от пациентов из Джибути (Восточная Африка), на основании филогенетического анализа была признана предположительным предшественником современных M.tuberculosis. Авторы полагают, что давние предшественники M.tuberculosis могли существовать 3 млн. лет назад и, возможно, микобактерии, дающие гладкие колонии, сосуществовали и распространялись с ранними гоминидами. Предложенный совместный с человеком эволюционный сценарий позволяет лучше объяснить степень генетического многообразия M.tuberculosis, наблюдаемого в различных регионах мира. Более того, эти исследования могут обеспечить дополнительное понимание эволюции специфических особенностей, значимых с точки зрения биологии и медицины, которые помогут лучше понять взаимодействие между возбудителем M.tuberculosis, популяцией хозяина и окружающей средой, что внесет существенный вклад в изучение туберкулеза.
Известно, что штаммовые кластеры и ОНП-группы продемонстрировали высокий уровень географической специфичности. Например, штаммы I и II групп преобладали среди пациентов Азиатского происхождения (включая Россию), и в то же время были минимально представлены среди штаммов южноамериканского происхождения.
Заключительная часть (5 мин)
Таким образом, анализ геномных различий между штаммами позволил по-новому взглянуть на филогенетическое родство между видами микобактерий туберкулезного комплекса. Было показано, что M.canettii на раннем этапе видообразования выделился в отдельную эволюционную ветвь и дивергировал от видов M.tuberculosis, M.bovis, M.microti, M.africanum и M.pinnipedii, при этом
M.bovis и M.tuberculosis расположены на разных ветвях филогенетического дерева, а не являются предковым и потомственным видами, соответственно.
Для выявления филогенетической структуры вида M.tuberculosis и определения родства между эпидемически значимыми штам-
РАЗДЕЛ 1. Лекционные материалы
173

МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
мовыми группами было использовано типирование по нсОНП, позволившее разделить M.tuberculosis на 3 ПГГ, которые дополнительно с использованием сОНП были разделены на 9 линий, внутри которых располагаются основные известные на сегодня штаммовые кластеры.
Рекомендуемая литература
1.Baker L., Brown T., Maiden M.C., Drobniewski F. Silent nucleotide polymorphisms and a phylogeny for Mycobacterium tuberculosis // Emerg. Infect. Dis. – 2004. – Vol. 10. – P. 1568–1577.
2.Brosch R., Gordon S.V., Marmiesse M., Brodin P., Buchrieser C., Eiglmeier K., Garnier T., Gutierrez C., Hewinson G., Kremer K., Parsons L.M., Pym A. S., Samper S., van Soolingen D., Cole S.T. A new evolutionary scenario for the Mycobacterium tuberculosis complex // Proc. Natl. Acad. Sci. USA. – 2002– Vol. 99. – P. 3684–3689.
3.Gutacker M. M., Smoot J. C., Lux Migliaccio C. A., Ricklefs S. M., Hua S., Cousins D. V., Graviss E. A., Shashkina E., Kreiswirth B., Musser J. M. Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: Resolution of genetic relationships among closely related microbial strains. // Genetics. – 2002. – V. 162. – P. 1533–1543.
4.Gutacker M. M., Mathema B., Soini H., Shashkina E., Kreiswirth B. N., Graviss E. A., Musser J. M. Single–Nucleotide Polymor- phism–Based Population Genetic Analysis of Mycobacterium tuberculosis Strains from 4 Geographic Sites // J. Infect. Dis. – 2006. – Vol. 193. – P. 121–128.
5.Gutierrez M.C., Brisse S., Brosch R., Fabre M., Omais B., Marmiesse M., Supply P., Vincent V. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis // PLoS Pathog. – 2005. – Vol. 1 (1). – e5.
6.Kapur V., Whittam T.S., Musser J.M. Is Mycobacterium tuberculosis 15,000 years old? // J. Infect. Dis. – 1994. – Vol. 170. – P. 1348–1349.
7.Sreevatsan S., Pan X., Stockbauer K.E., Connell N.D., Kreiswirth B.N., Whittam T.S., Musser J.M. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination // Proc. Natl. Acad. Sci. USA. – 1997. – Vol. 94. – P. 9869–9874.
Материально-техническое обеспечение
Мультимедийная или проекционная демонстрационная системы, экран, лазерная указка; набор тематических слайдов.
174

Тема 8.4. «Молекулярная эпидемиология туберкулеза в России»
Количество аудиторных часов – 1
Примерный план лекции
Основные вопросы, освещаемые в лекции:
•Кластерный анализ популяции МБТ, циркулирующей в регионах РФ с использованием сполиготипирования
•Кластерный анализ по ПДРФ IS6110
•Генотипирование МБТ, выделенных на территории РФ, методом VNTR и MIRU-VNTR
Вводная часть (5–10 мин)
Туберкулез в Российской Федерации представляет собой большую медико-социальную проблему. Статистический анализ заболеваемости ТБ в 2011 г. выявил 73 новых случая заболевания туберкулезом на 100 000 населения. Смертность от ТБ была зафиксирована у 14,2 на 100 000 населения. Отдельную проблему представляет крайне высокий уровень распространения штаммов с МЛУ с его неуклонным нарастанием. В 2011 году отмечено 15,5% случаев МЛУ туберкулеза среди новых случаев заболевания туберкулезом, и 34,2% среди ранее леченых.
Понимание эпидемиологии туберкулеза особенно важно, поскольку является основой для разработки мер профилактики, диагностики, лечения и программ борьбы с туберкулезом для органов здравоохранения. Применение методов молекулярной эпидемиологии для типирования микобактериальных штаммов внесло существенный вклад в исследование трансмиссии туберкулеза.
Основная часть (30 мин)
На сегодня в мире существует три основные методики молекулярного типирования штаммов МБТ – по ПДРФ IS6110, по особенностям строения региона прямых повторов (сполиготипирование) и по числу тандемных повторов.
Анализ циркулирующей в РФ популяции штаммов МБТ с использованием типирования по ПДРФ IS6110. Генотипирование штаммов по ПДРФ IS6110 основано на числе и расположении IS6110 в геноме МБТ. Исследования методом ПДРФ IS6110 проводились в Центральном, Южном, Приволжском, Сибирском федеральных округах на 1227 штаммах МБТ. Было выявлено более 42 кластеров, из них преобладали два – W и AI, однако доминирующим был W (более 40%).
РАЗДЕЛ 1. Лекционные материалы
175
|
При анализе этим методом культур, выделенных от больных в |
||
|
|||
|
Центральном федеральном округе, было показано, что МБТ кла- |
||
|
стеров W и AI встречались приблизительно в равных соотноше- |
||
|
ниях (33% – W и 26% – AI). |
||
|
Похожее распределение МБТ двух основных генотипов наблю- |
||
|
далось у больных в Приволжском федеральном округе. |
||
|
Исследование, проведенное на мигрантах из Южного феде- |
||
|
рального округа, показало очень высокую встречаемость МБТ W |
||
|
кластера – 62,0%. МБТ AI кластера составили лишь 14,1%. |
||
|
В Сибирском федеральном округе также выявлялся большой |
||
|
процент МБТ W кластера (более 50%), а в Томской области были |
||
|
выявлены штаммы кластера KY, эндемичные для этого региона. |
||
|
Таким образом, генотипирование штаммов по ПДРФ IS6110 |
||
|
продемонстрировало преобладание в России МБТ двух основных |
||
|
кластерных групп – W и AI, встречаемость которых варьировала в |
||
|
зависимости от региона, однако преобладающим всегда оставался |
||
|
кластер W и частота его встречаемости могла достигать более, чем |
||
|
70%. |
|
|
|
Анализ циркулирующей в РФ популяции штаммов МБТ с исполь- |
||
|
зованием сполиготипирования. Метод сполиготипирования основан |
||
|
на анализе локуса прямых повторов микобактериального генома. |
||
|
Этот участок характеризуется наличием многократно повторяю- |
||
|
щихся участков с одинаковой последовательностью (прямые по- |
||
|
вторы), между которыми расположены спейсорные последова- |
||
|
тельности, отличающиеся между собой по длине и нуклеотидному |
||
|
составу. |
||
|
Методом сполиготипирования проведены исследования в |
||
|
Центральном (Московская область, Ивановская область, Тульская |
||
|
область), Северо-Западном (Ленинградская область, Республика |
||
|
Карелия, Архангельская область, Калининградская область), При- |
||
РЕКОМЕНДАЦИИ |
волжском (Самарская область) и Сибирском (Новосибирская |
||
область, Республика Тыва) федеральных округах Российской Фе- |
|||
|
|||
|
дерации. |
||
|
В результате сполиготипирования было описано более 6 основ- |
||
|
ных кластеров (согласно базы данных SpolDB3), из которых пре- |
||
|
обладали МБТ группы Beijing. Особенностью МБТ данной группы |
||
МЕТОДИЧЕСКИЕ |
является наличие в DR-регионе всего 9-ти спейсорных последо- |
||
вательностей из 43-х. Преобладание данного кластера было пока- |
|||
|
|||
|
зано во всех исследованных территориях и варьировало от 34% до |
||
|
74% |
|
|
|
|
|
|
176

Этот факт существенно отличает Российскую популяцию штаммов МБТ от Европейской. В Европе не выявлено преобладающего сполиготипа МБТ, однако показано, что МБТ кластера LAM встречаются в 9% случаев, Haarlem – 8%, а Beijing – лишь в 4% случаев. Однако для РФ прослеживается сходство сполиготипов циркулирующих штаммов с Ю-В Азией, популяция которых характеризуется преобладанием именно Beijing генотипа.
Анализ циркулирующей в РФ популяции штаммов МБТ с использованием типирования MIRU-VNTR. VNTR типирование по 5 локусам показало, что среди 245 штаммов, выделенных от больных из Москвы и из Самарской области, было выявлено более
20VNTR-типов и преобладал 42425 – около 50% от всех штаммов. Анализ числа тандемных повторов в 24 локусах (MIRU-VNTR),
выполненный для 115 штаммов M.tuberculosis, выделенных от больных из Центрального региона Российской Федерации, выявил 71 различных VNTR-профилей, из которых 59 (83,0 %) были уникальными (обнаруживались только у одного штамма в выборке), 56 штам мов M.tuberculosis образовывали 12 кластеров, включающих в себя от 2 до 13 штаммов. Три основных кластера, содержащих 13 (11,3 %), 12 (10,4%) и 9 (7,8%) штаммов M.tuberculosis имели MIRU-VNTR- профили 223325173533424454433672, 223325153533424454433682 и 223325135334244544336622, соответственно.
Такое распределение соответствует представленным ранее данным по Северо-Западному региону (Санкт-Петербург), Центральному региону (Самарская обл.), Уральскому региону и, вероятно, характерно для России в целом. Похожее распределение наблюдается и в некоторых странах Азиатского региона, в частности, в Китае.
Таким образом, анализ генотипов МБТ, циркулирующих на территории Российской Федерации, проведенный по трем основным генетическим маркерам, показал преобладание штаммов со сходными генетическими характеристиками: W генотип согласно типированию по ПДРФ IS6110, Beijing сполиготипа и VNTR-типа 42425.
Известно, что штаммы W-Beijing часто ассоциируются с МЛУ. Анализ 164 МЛУ штаммов и 180 чувствительных штаммов M.tuberculosis показал, что 49% МЛУ штаммов принадлежали к группе W-Beijing. Среди чувствительных штаммов МБТ этой группы встречались лишь в 24% случаев.
РАЗДЕЛ 1. Лекционные материалы
177