

|
1. Molecular mechanics calculations |
51 |
|
|
R |
R |
|
|
Pri |
Pri |
|
|
N |
N |
|
H |
H |
H |
H |
|
Pri |
Pri |
|
|
G+ |
G− |
|
|
|
R |
|
|
Pri |
Pri |
|
|
|
N |
|
|
H |
H |
|
A
FIGURE 4. Newman projections (along the N CH2 bond) for the three rotamers of RCH2 N (i-Pr)2
ButCH2 N Pri |
ButCH2 N But |
Pri |
CH2 CH3 |
(38) |
(40) |
around single bonds. Further cooling to 108 K slows down the low-energy barrier process and gives rise to 2 signals of 9:1 ratio in the 13C-NMR spectra. Assuming that the only relevant structures of 40b are those with anti t-butyl groups, MM2-85 was used to calculate the three possible rotamers around the N ethyl bond (40c e). On the basis of the relative MM2 steric energies (40c: 0.00; 40d: 1.20; 40e: 0.67 kcal mol 1), and assuming identical entropies, the relative populations of the three rotamers were calculated to be 64.9%:8.6%:26.5% at 298 K and 95.2%:0.4%:4.4% at 108 K. The good agreement with the above NMR results at 108 K (peak ratio of 9:1) suggests that the low barrier process indeed relates to N ethyl rotation. In addition to the energy minima, the complete rotational potential around this C N bond was calculated and is presented in Figure 5. The barrier to the 40c ! f40dg ! 40e interconversion is 5.7 kcal mol 1, in very good agreement with the experimental result (6 kcal mol 1).
|
|
|
N-inversion |
But |
|
t |
N |
Bu |
t |
rotation |
|
N |
Et Bu |
But |
Et |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
(40a) |
|
|
|
|
(40b) |
|

52 |
|
Pinchas Aped and Hanoch Senderowitz |
|||||
|
) |
10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
−1 |
|
|
|
|
|
|
|
mol |
8 |
|
|
|
|
|
|
(kcal |
|
|
|
|
|
|
|
6 |
|
|
|
|
|
|
|
energy |
|
|
|
|
|
|
|
4 |
|
|
|
|
|
|
|
strain |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Relative |
2 |
|
|
40c |
40d |
|
|
|
40e |
|
|
|
|
|
|
|
0 |
|
|
|
|
|
|
|
180 |
120 |
60 |
0 |
−60 |
−120 |
|
|
|
Me-C-N-lone pair dehedral angle (deg) |
||||
40e
−180
FIGURE 5. Rotational energy profile about the N-ethyl bond for N-ethyl-N-t-butylneopentylamine (ETNA, 40) as calculated by MM2-85. Reproduced with permission from Reference 74
H3 C |
|
|
|
|
|
H |
CH3 |
But |
|
But |
CH But |
H |
H |
|
|
|
2 |
||
|
CH2 But |
|
|
|
|
H |
|
(40c) |
|
(40d) |
|
|
H |
H |
|
|
But |
CH2 But |
|
|
|
|
|
|
|
CH3 |
|
|
|
(40e) |
|
Tertiary amines of |
class (iii) were |
also studied by Lunazzi and |
coworkers75. A |
series of beta-substituted-alpha-amino naphthalenes 41a d 44a d were investigated using DNMR and NOE experiments and molecular mechanics. Such systems allow for separation between rotational processes, whose barriers around Ar N bonds are quite high (15 23 kcal mol 1), and inversion, known to have particularly low barriers

1. Molecular mechanics calculations |
53 |
in aromatic amines ( G‡ D 7.24 kcal mol 1 for aniline76). The systematic increase in
measured G‡ values with the bulkiness of R (on going from a to d) supports the exclusion of N-inversion from this dynamic process. (As stated before, this conclusion is rather questionable, and the DNMR detected process may better be characterized as an inversion rotation type). Detailed MM2-82 calculations for the N,N-diethyl structures 42a and 42d show 4 energy minima, all having the inversion plane approximately perpendicular to the naphthalene plane, in agreement with the NMR conclusions (Scheme 4). (A recent structural investigation of crowded tertiary amines77 supports this finding: While dimethylaniline was found to adopt a (minimum energy) conformation in which the lp is approximately perpendicular to the plane of the phenyl ring, the more hindered N-(2,6-diisopropylphenyl)-piperidine adopts a 90° twisted conformation (NMR, X-ray performed on the meta-nitro derivative). An intermediate situation was calculated (MM2) for the parent N-phenylpiperidine system.) The analysis of the conformational space was performed by a combination of NOE and molecular mechanics, the latter used to establish the various interproton distances. The quasi-planar structures of 42a d were also calculated, to estimate the transition state energies. The observed trend is well reproduced by the calculations giving (MM2 results in parentheses) 17.5 (12.0), 18.6 (16.0), 19.3 (17.1) and 21.2 (22.3) kcal mol 1 for 42a d, respectively, with an average deviation of about 15%.
N
R
I |
II |
|
|
R |
R |
E = 51.3 |
E = 51.6 |
III |
IV |
|
|
R |
R |
E = 51.4 |
|
E = 51.9 |
SCHEME 4. Top view of the conformers (I |
|
IV) corresponding to the four energy minima (E, |
|
kcal mol 1) as obtained by MM2 calculations for 42a (R D Me) and 42d. Reproduced with permission from Reference 75

54 |
Pinchas Aped and Hanoch Senderowitz |
|
|
Me |
Et |
Et |
Et |
|
N |
|
N |
|
R (41a-d) |
(42a-d) |
R |
|
a = Me |
a = Me |
|
|
b = Et |
b = Et |
|
|
c = Pri |
c = Pri |
|
|
d = But |
d = But |
|
Me Pri
N
R (43a-d) |
(44a-d) |
a = Me |
a = Me |
b = Et |
b = Et |
c = Pri |
c = Pri |
d = But |
d = But |
Et Pri
N
R
Cyclic tertiary amines are characterized by a smaller number of dynamical processes, in comparison with the acyclic analogs, due to ring constrains. Thus, sterically hindered 2,2,6,6-tetramethyl piperidines (45 51) were subjected to DNMR analysis combined with MM3-92 calculations67a. The calculations included all possible rotamers based on the chair conformation of the piperidine ring, except for the N-isobutyl derivative 48, where the twist-boat forms were also considered. The computed structures of the energy minima are in accord with experimentally observed phenomena. For example, the presence of intramolecular hydrogen bond, indicated by both NMR and IR spectra of 50 and 51, is accounted for by the calculated structures of the corresponding minima with an OH. . .N distance of ca 2.1 A˚ . Similarly, the flattening of the crowded amino group, detected in the X-ray structure of 1,2,2,6,6-pentamethyl-4-tert-butyl-4-hydroxypiperidine (C2 N C6
angle of 118.2°78 ), is also well reproduced (C2 N C6 D 117° 118.5° for compounds 45 51). The ‘double driver’ option was used to map the potential energy surface of compounds 46 50 in terms of the N C˛ and C˛ Cˇ torsions and to locate minimumenergy conformers and lowest-energy pathways among them. The calculated results for the rotational barriers are in good agreement with the experimental ones [DNMR (MM3): 10.6 (9.0), 10.9 (9.6), 12.4 (11.9), 16.7 (13.8), 18.5 (17.9)], where MM3 underestimates the barriers by an average of 1.3 kcal mol 1, a standard deviation of less than 12%.
45 |
46 |
47 |
48 |
49 |
50 |
51 |
||||
N |
|
|
|
|
|
|
|
|
|
|
R = Me |
Et |
Bu |
But |
|
CH2 |
|
C(R1) (R2) CH3 |
|||
|
|
|
||||||||
|
|
|||||||||
R |
|
|
|
|
|
|
|
|
|
|
(45-51) |
R1 = |
|
|
|
|
Me |
Me |
Ph |
||
|
|
|
|
|
|
|
|
|
|
|
|
R2 = |
|
|
|
|
OAc |
OH OH |
|||
|
|
|
|
|
|
|
|
|
|
|
Unlike the MM2 force field, there is no explicit treatment of the nitrogen (or oxygen) lone pair in MM3. This allows one to calculate planar amine structures, as a model to

1. Molecular mechanics calculations |
55 |
transition structures for nitrogen inversion. This procedure, however, should be practiced with caution since the force field has been parameterized for pyramidal sp3 N type amines. Belostotskii and coworkers79 have recently taken advantage of this feature and estimated the inversion barriers in a series of rigid, hindered cyclic amines (52a f, 53 58). The calculated results (Table 24), generally show the correct trend, and reproduce about 85% (standard deviation) of the experimental G‡ values.
R
N
a |
b c |
d |
e |
f |
|
|
|
|
|
R = Me Et Pri Bu Bui But
(52a-f)
Me |
N |
Me |
N |
Me |
N |
|
|
||||
|
|
|
|
|
(53) |
|
(54) |
(55) |
|
N |
Me |
Me |
N |
Me |
|
|
|||
N
(56) |
(57) |
(58) |
2.Polyamines
a. Diamines. The simplest diamine, methylenediamine, belongs to a more general type of compound with an X C Y moiety, which is characterized by the anomeric effect. The molecular mechanics treatment of such structures has been described in Section II.B.5 of this review. Other diamines, in which the amino groups are separated by more than
one carbon atom, were studied by the CFF81, MM2-8782 and MM3 force fields (see Section II.C.2 for the latter case). The conformational space of 1,2-ethanediamine (EDA) can be defined by the two peripheral lp N C C, and the central N C C N dihedral angles. Selected energetic and geometrical results for the lowest-energy conformers of

56 Pinchas Aped and Hanoch Senderowitz
TABLE 24. Experimental (DNMR) and calculated (MM3) inversion barriers of several azabicyclo and azatricyclo compounds
|
G‡ [Reference] |
MM3 estimated barrier |
|
Compound |
to inversion (kcal mol 1) |
||
52a |
13.8 |
[80a] |
10.6 |
52b |
6.1 |
[79] |
4.9 |
53 |
8.2 |
[80b] |
8.3 |
54 |
7.1 |
[80c] |
6.6 |
55 |
5.7 |
[80d] |
4.3 |
56 |
7.8 |
[80c] |
6.6 |
57 |
8.8 |
[80e] |
8.0 |
58 |
14.4 |
[80a] |
10.0 |
|
|
|
|
TABLE 25. Selected energetic and geometrical results for 1,2-ethanediamine (EDA) as calculated ab initio and by the CFF and MM2 force fields and obtained experimentally (relative energy in kcal mol 1, bond lengths in A,˚ bond angles and torsional angles in degrees)
Conformera |
Method |
Erel |
C C |
C N |
N C C lp N C C |
N C C N C C N lp |
||||
AGG0 |
MM282 |
0.00 |
1.536 |
1.456 |
111.5 |
178.6 |
62.9 |
|
62.0 |
|
|
CFF |
81 |
0.01 b |
1.548 |
1.475 |
110.6 |
180.0 |
60.8 |
|
|
|
|
61.9 |
||||||||
|
|
|
(0.00) |
|
|
|
|
|
|
|
|
ab initioc |
0.04 |
1.539 |
1.475 |
111.0 |
|
57.8 |
|
|
|
|
|
|
(0.00) |
|
|
|
|
|
|
|
|
MW83 |
|
1.546 |
1.469 |
111.5 |
|
63.0 |
|
|
|
GGG0 |
MM2 |
0.39 |
1.536 |
1.457 |
111.3 |
66.3 |
66.2 |
59.1 |
||
|
CFF |
|
0.03 |
1.547 |
1.475 |
110.4 |
63.1 |
61.9 |
61.9 |
|
|
|
|
(0.02) |
|
|
|
|
|
|
|
|
ab initio |
0.00 |
1.532 |
1.474 |
108.6 |
|
61.9 |
|
|
|
|
|
|
(0.50) |
|
|
|
|
|
|
|
|
MW |
|
1.546 |
1.469 |
109.0 |
|
63.0 |
|
|
|
|
ED84 |
|
1.545 |
1.469 |
110.2 |
|
64.0 |
|
|
|
aA D 180°, G D 60°, G0 D 60°.
bIn parentheses, relative free enthalpies corrected for statistical weights. CFF calculates another conformer, AAG, as the global minimum; see text.
c3-21G calculations from Reference 85. In parentheses, 6-31G relative energies from Reference 81.
EDA, as obtained by molecular mechanics and ab initio calculations, together with gasphase experimental data, are presented in Table 25. Electron diffraction suggests that the most stable form of EDA has a gauche central torsional angle, while the microwave results were interpreted in terms of two stable conformers, AGG0 and GGG0 . While both MM2 and ab initio calculations confirm these findings, CFF locates another lowenergy conformer, AAG, and calculates all three to be within 0.02 kcal mol 1 (enthalpies corrected for statistical weights) of the global minimum. The CFF force field seems to overestimate the stability of conformations with an anti N C C N torsion (all are calculated to be at least 1.3 and 1.1 kcal mol 1 above the lowest-energy form by MM2 and ab initio, respectively). This is attributed to the lack of hydrogen-bond interactions in this version of the force field. The geometrical features, though, are reproduced somewhat better than by MM2.
The next linear diamine, 1,3-propanediamine (PDA), was also studied by the same force fields. Four conformational families were considered and defined by the two

1. Molecular mechanics calculations |
57 |
central N C C C torsions, while the two peripheral lp N C C angles determined the conformations within each group. In the lack of experimental data for PDA, the only reference for comparison came from partial 6-31G ab initio results of one representative form of each group81, which indicated that the two most stable conformers belong to the G0 A and AA conformational families. CFF calculations of the entire conformational space indeed found the lowest conformers of each of these groups to be among the four most stable forms, within only 0.01 kcal mol 1 (corrected enthalpies, see above) of the global minimum. All conformers belonging to the other two groups, namely G0G0 and G0 G, were calculated to be at least 0.4 and 0.6 kcal mol 1 higher in energy. MM2 reversed the order of the two lowest groups, but only by 0.7 kcal mol 1, and calculated the other two to be at least 1.24 and 1.42 kcal mol 1 above the global minimum.
A series of alkyl substituted 1,4-diazacyclohexanes (piperazines) was also studied by MM282. These were divided into three categories according to the degree of substitution on the nitrogen atom (0, 1 and 2 alkyl substitutions) and their conformers were defined by the substituent orientation at the carbon and nitrogen centers (axial or equatorial). The few experimental data, relevant to piperazine conformational analysis, consist of dipole moment measurements86, ultrasonic relaxation spectra87 and Raman spectroscopy88, all measured in solution. A comparison between the MM2 calculated conformational ratios and those obtained experimentally does not lead to a decisive conclusion. The discrepancies were attributed89 to the questionable reliability of the dipole moment measurement technique on the one hand, and to the possible role of intermolecular hydrogen bonds which were not accounted for in the calculations, on the other. Similarly, barriers to ring inversion, in the range of 9.6 20.6 kcal mol 1, are only qualitatively reconstructed. Again, the experimental data fail to provide a definite reference point. For example, values in the range of 12.2 16.2 kcal mol 1 were published for the lowest barrier of N,N0- dimethylpiperazine90. MM2 calculations faithfully reproduce the ED deduced geometries of the unsubstituted piperazine and its N,N0-dimethyl derivative. Most bond lengths and angles are within the experimental error, except for the ring C N C angle in the latter compound, for which the difference exceeds 4°.
MM2 calculations (a PC version of MM2-89) were used in a theoretical conformational analysis of substituted ethanes with vicinal phenyl groups of the general form R1PhCH CHPhR2 where R1 and R2 are amino and/or hydroxy moieties91. Conformational stability in these systems results from the interplay between three major effects: (1) steric interactions between the neighboring phenyl groups and the R1 and R2 substituents; (2) electrostatic interaction between the phenyl groups; (3) intramolecular O H. . .O, O H. . .N, N H. . .O and N H. . .N hydrogen bonds. The inclusion of hydrogen bonding parameters in MM230a allowed for the treatment of the latter interaction type, while the unique electrostatic effect was accounted for through the V1 parameter of the C(sp2) C(sp3) C(sp3) C(sp2) torsional potential. The conformational space for these compounds was defined by the three possible relative orientations of the phenyl groups about the Ph C C Ph torsion, i.e. ap (antiperiplanar), (C)-sc (Csynclinal) and ( )-sc ( synclinal). Comparison with experiment was performed for the vicinal coupling
constants between the two benzylic hydrogens. 3Jgauche and 3Janti were calculated for the lowest-energy minima of the main three conformational families (using a Karplus-type
equation92), averaged over the corresponding MM2 calculated populations and compared with the NMR results. A fairly good correlation was obtained between the computed and experimental values, with an average deviation of 1.0 Hz for a total of 20 comparisons.
b. Triand tetraamines. The smallest aliphatic triamine, propane-1,2,3-triamine (PTA, 59), was calculated with a reparameterized MM2 force field93. The only source of

58 |
Pinchas Aped and Hanoch Senderowitz |
experimental data for comparison came from an X-ray structure of the trihydrochloride monohydrate salt, and the parameterization was aimed at fitting this structure by changing the values of the appropriate stretching and bending parameters for the amino and ammonium nitrogen. The resulting new parameter set resembled that used for cobalt complexes of triamines94. (Ammonium compounds, as well as amino-metal complexes, are generally beyond the scope of this chapter, and are only briefly mentioned in connection with their neutral, or metal-free, forms.) Atomic charges for this system came from MNDO calculations (the authors used a MacroModel implementation of MM2, with atomic charges rather than bond dipoles), and the neutral form of PTA as well as its mono-, diand tri-protonated derivatives were conformationally analyzed. The conformational map of PTA may be defined via the two C C C N dihedral angles, giving rise, in principle, to six basic forms (regardless of the nitrogen lone-pair orientations), namely AA, AGC (G A), AG (GC A), GC GC (G G ), GC G and G GC . The most stable conformation was calculated to be the AA one, though this could not be confirmed experimentally. For the tri-protonated derivative, however, the AA form was found to be only the third best, 3.13 kcal mol 1 above the global minimum. The two most stable calculated conformers, separated by only 0.27 kcal mol 1, were of the AGC and AG types, and closely resembled the structures of two PTA.H33C cations found in the asymmetric unit.
N |
N |
|
H |
|
|
C2 |
N |
N |
|
C1 C3
|
NH2 |
N |
N |
|
|
NH2 |
N2 NH2 |
|
|
N |
N |
N1 |
N3 |
|
|
||
|
(59) |
(60) |
|
|
(61) |
|
N |
N |
N |
N |
|
|
N |
N |
N |
N |
|
|
(62) |
|
|
(63) |
|
Small and medium-size macrocycles bearing four nitrogen atoms were calculated95 with an early force field used in the EFF program96,97 as part of a study dealing with the extra stability of metal complexes formed by the cyclic ligands over their open-chain analogs. Thus, the structures of 1,4,8,11-tetraazacyclotetradecane (cyclam 60) and 2,3,2- tet(1,4,8,11-tetraazaundecane), 61, as well as their Ni(II) complexes, were optimized. The observed differences in enthalpy upon complex formation, between the cyclic (62 and 60) and open chain (63 and 61, respectively) ligands, could only be reproduced when a N Ni bond involving a secondary nitrogen was made 1.7 kcal mol 1 more stable than a similar
1. Molecular mechanics calculations |
59 |
bond to a primary nitrogen. Hence the authors concluded that the above extra stability, known as ‘the macrocyclic effect’ and observed also for smaller rings, could be attributed to the presence of more secondary nitrogens in the cyclic analog. A potential problem lies in the values of the bond dipoles used in these calculations which, although considered unreliable in EFF98, still had a significant influence on the calculated H: For the openchain compounds, where the dipoles were free to rotate to the most favorable orientation, the total energy was relatively independent of their values. In the cyclic compounds, however, the dipoles were ‘forced’ into close proximity in the central ‘hole’, giving rise to considerable electrostatic repulsion, which would be relieved on complex formation. Since this effect is much more pronounced in the gas phase than in aqueous solutions, where the dipoles are solvated by water molecules, the authors chose to use much smaller bond-dipole values.
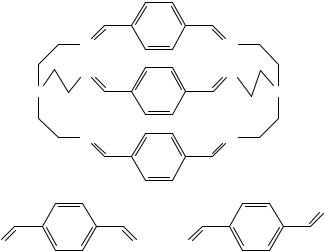
c. Cryptands and azacrown ethers. Several examples are found in the literature where molecular mechanics calculations have been used to explore the structure and conformational space of host molecules of the cryptand99,100 and azacrown101 families. The two known crystal structures of the hexaimino cryptand 64100 were used to derive four starting conformations. These were denoted as SSS, SSO, SOO and OOO, where S and O stand for syn and anti arrangements of the iminic nitrogen atoms around each phenyl group (see Figure 6). A molecular dynamics based conformational search was used to locate all possible arrangements of the four basic conformers, and these were later minimized using the MM2MX program102. Several parameters had to be added to the force field, for example the NDC C(Ar) C(Ar) and NDC C(Ar) H torsional V2 parameter (fitted by the authors) and several bond dipoles which were taken from MM2-87 or derived from MOPAC calculated atomic charges. The specific semiempirical method used to derive the bond dipoles was not given in References 100 and 101. The latter parameters seem to have a significant influence on the relative stability of the above four conformers.
N |
N |
N |
N |
N |
N |
N |
N |
N
N N N
S O
FIGURE 6. Schematic representation of the hexaimino cryptand (64)

60 |
Pinchas Aped and Hanoch Senderowitz |
MOPAC derived bond dipoles were also used by Santos and Drew101 to reparameterize MM2-87 for a conformational analysis study of 18-azacrown-6, 65. Seven starting conformations were taken from previous calculations of the oxygen analog 18-crown-6, together with one observed in two crystal structures containing the [H6(18-aza-crown)]6C cation. Several series of calculations were performed to test the effect of various bond-dipole values, and the inclusion/exclusion of lone pairs on the nitrogen atoms. As long as lone pairs were included, the highest symmetry, D3d, conformer was found to be the global minimum, regardless of the bond-dipole values. The energetic preference of this form over the next best conformations varied though, between 1.7 kcal mol 1 for structures minimized using half the MOPAC values, to 4.0 kcal mol 1 when using the full bond dipoles. When no nitrogen lone pairs were included, the Ci, C0i and C2 forms became more favorable. These results significantly differ from those of the oxygen analog, for which the Ci form was found to be the most stable one, both by molecular mechanics calculations (AMBER103a,b, VBFF103c and MM2103d) and also as the conformation adopted in the crystalline state104. Only when no bond dipoles or atomic charges were assigned to 18-crown-6 (which seems highly unrealistic) did the D3d form come out as the most stable one. MOPAC calculations of 18-azacrown-6 largely agree with the MM2 results. The molecular mechanics minimized structures were later used as starting geometries for calculations of the H33C protonated macrocycle, as well as for its host guest complexes with small organic anions.
N N
N N
N N
(65)
3. Medium-size rings
An extensive study of the conformational behavior of perhydroazepine 66 was carried out by Espinosa and coworkers105. This pure force field based analysis, with no reference to experiment or other theoretical methods, was part of a series of studies on cycloheptane, cycloheptene and some of their oxygen, sulfur and nitrogen heterocyclic analogs. MM2
H
N
(66)