

1. Molecular mechanics calculations |
71 |
||||||||
R |
N |
R′ |
|
R′ |
|
|
|
R′ |
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
R |
|
|
R |
|
|
|
N |
|
|
|
|
|
|
||
O |
|
|
|
|
N |
|
|
|
|
|
|
|
|
O |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
R |
R′ |
|
|
|
R |
R′ |
|
|
|
|
|
|
|
|
|
|
|
|
(87) |
Me |
Me |
(90) |
|
H |
H |
|
||
(88) |
Et |
Et |
(91) |
|
Me |
H |
|
||
(89) |
Pri |
Pri |
(92) |
|
Me |
Me |
|
||
(93)Ph Ph
(95) Ph Me
N
N
O
(94)
one group of aromatic (93 95) N-nitrosamine derivatives were studied. The downfield shift observed in the 17O spectrum when going from the aliphatic to the aromatic compounds was larger than for the corresponding amides (C55 ppm difference between 87 and 95, compared with only C26 ppm between N,N-dimethylformamide and N-methyl- N-phenylacetamide). This is in accord with the MM2 calculated torsional angle between the phenyl plane and the functional group (nitroso or carbonyl) for the two systems, namely 62° for the amide120 but only 23° for compound 95, allowing for a much better conjugation. The latter value is also in good agreement with the microwave result for
this system (28°121 ). MM2 calculates two different torsional angles, 30° and 47°, for the diphenyl derivative 93, and a value near 0° for the rigid carbazole system of 94. These values are in accord with the NMR results, which show a C93 ppm downfield shift when going from 95 to 94, but only about a third of this value between 95 and 93. A smaller diversity exists among the aliphatic compounds with 5 and 9 ppm ranges for 87 89 and 90 91, respectively (an exception to the above is found for compound 92 which is further shifted by C67 ppm, relative to 91). In contrast, the 15N chemical shifts seem to be determined mostly by steric factors both for N-1 and N-2 ( 39.5/C9.7 ppm shifts for N-1/N-2, on going from 87 to 89, but only 36.9/C9.6 when comparing 87 with 93). The steric dependence of the 17O chemical shift may be well explained by the MM2 optimized structures of 87 92: In all cases save 92, the nitroso oxygen lies near a C-alpha hydrogen, thus experiencing similar environments, and the nitroso group is calculated to be coplanar with the average plane of the ring (C N N O torsional angle ranging from 22° to 27°). Only in 92 is the nitroso group forced into close proximity with the methyl group. Indeed, a linear relationship is found between the differences in MM2-estimated VdW energies
72 |
Pinchas Aped and Hanoch Senderowitz |
and the changes in chemical shifts for 90 92 (though a definite conclusion should not be drawn, of course, from three points only).
C. Mechanisms of Chemical Reactions
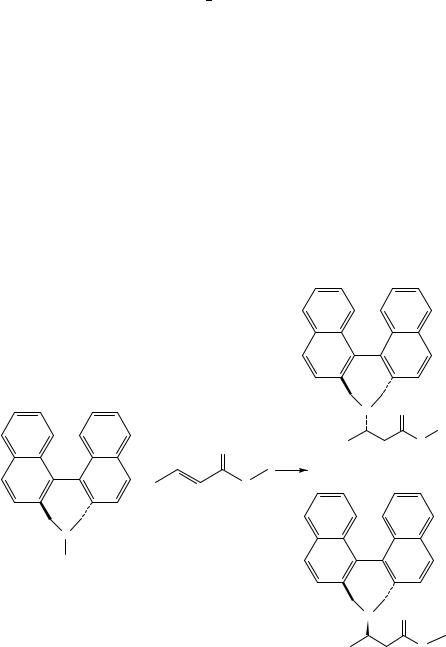
Following their molecular-mechanics-based study on intramolecular radical additions in hydrocarbons122 Houk’s group have used a slightly modified MM2 force field to model the stereoselective Michael addition of an optically active amine to methylacrylate123. When the reaction employs the neutral amine 96, a ratio of 4.1:1 for the diastereomers 98 and 99 is obtained as the kinetic product, followed by a slow equilibration of the reaction mixture to a 1:1 ratio (Scheme 7). The corresponding lithium amide, however, gave isomer 99 almost exclusively. The model developed for the addition to the neutral amine assumes a tetrahedral N. . .C(sp2) C(sp2) attack angle, and a 2.0 A˚ N- - -C transition state bond length. This latter parameter has been introduced into the MM2 force field as a special ‘natural’ bond length between the nitrogen and the olefinic carbon, beta to the ester carbonyl. The torsional potential around this bond was calculated using various values for the H N. . .C(sp2) C(sp2) dihedral angle, and the resulting curves, for both 98- and 99-like transition states, denoted as 98a and 99a, are presented in Figure 10. These torsional potentials show two clear local minima for each case: near 150° and 330° for 98a, and at about 60° and 250° for 99a. A Boltzmann distribution, calculated from
N O
O
|
O |
|
(98) |
+ |
O |
|
(97) |
N
R
(96)
N O
O
(99)
SCHEME 7

1. Molecular mechanics calculations |
73 |
Relative energy (kcal mol−1)
6.0
5.0
4.0
3.0
2.0
1.0
0.0
0
|
H |
H |
|
|
|
|
CH3 |
|
|
|
|
|
Naph. |
Naph. |
|
|
|
|
O |
|
|
|
|
|
O |
|
|
|
|
|
|
99a |
|
|
|
|
H |
H |
|
|
|
|
H3C |
|
|
|
|
|
Naph. |
Naph. |
|
|
|
|
|
O |
|
|
|
|
O |
|
|
|
|
|
98a |
|
|
|
|
60 |
120 |
180 |
240 |
300 |
360 |
H-N...C(SP2)-C(SP2) dihedral angle (deg)
FIGURE 10. The torsional potential (relative energies) around the N. . .C bond in the two model transition state structures, 98a and 99a, for the neutral Micheal addition
the molecular mechanics energies of these minima, predicts a 4:1 product ratio, in very good agreement with the observed kinetic product mixture. Since the two diastereomeric products are calculated to have nearly identical steric energies, the final thermodynamic ratio of 1:1 can also be rationalized. For the ionic reaction, a simple model of a six-center transition state involving a H(N). . .ODC interaction was assumed (structures 100 and 101), where the H(N) simulates the Li cation. This cyclic structure fixes the transition state to H N. . .C(sp2) C(sp2) values close to 300° and 60°, for the 98a- and 99a-like species, respectively, making 101 more stable, and leading to the kinetic predominance of product 99.
|
O |
O |
O |
O |
|
|
|
||
|
H |
|
|
H |
H |
|
|
|
H |
Naph. |
|
Naph. |
Naph. |
Naph. |
|
CH3 |
|
|
CH3 |
|
(100) |
|
|
(101) |
In another work, Maryanoff and Almond have used a combined molecular mechanics experimental approach to rationalize the high 1,4-diastereoselectivity in the hydrogenation/reduction of hexahydro-1-phenyl-3-benzazonine derivatives and the NMR spectra and relative stability of the resulting diastereomers124. Two diastereomers with a nitrogen containing a nine-membered ring, 103 and 105, were synthesized by hydrogenation and reduction of the appropriate precursors, 102 and 104, respectively (Scheme 8). Upon treatment with a basic solution, amine 103 was completely converted

74 |
Pinchas Aped and Hanoch Senderowitz |
|||
|
Ph |
Me |
Ph |
|
|
|
N |
|
|
|
|
|
|
H2 /Pt |
|
|
|
|
Me |
|
(102) |
|
|
(103) |
|
|
|
|
OH − |
|
Ph |
|
|
Ph |
|
|
|
|
|
|
|
Me |
Na |
|
|
|
|
||
|
+ |
I |
− |
NH3 |
|
N |
|
|
|
|
Me |
|
|
Me |
|
|
|
|
|
|
(104) |
|
|
(105) |
|
|
|
|
SCHEME 8 |
Me
N
Me
N
into a 3:7 ratio mixture of its diastereomeric counterpart, 105, and an elimination product (not shown in Scheme 8). Molecular mechanics calculations were performed with three main objectives: (1) assigning the stereochemistry of the two hydrogenation/reduction products (103 and 105), which could not be done based on the NMR spectra only;
(2) rationalizing the differences in the thermodynamic stability of diastereomers 103 and 105, as suggested by the disappearance of the former and the persistence of the latter under basic conditions (see above); (3) suggesting a model to account for the diastereofacial selectivity in the catalytic hydrogenation 102 ! 103. The conformational space of the nine-membered ring was explored as follows: Initial geometries were obtained from the four stable conformations of cyclononane found by Anet and Krane125. These were searched for reasonably flat dihedral angles and, wherever such an angle was found, a benzo ring was fused onto the cyclononane. The initial set of starting geometries (18) was doubled by adding the appropriate substituents at both axial and equatorial positions. MM2 minimization of the 36 conformations obtained for each diastereomer gave, after elimination of all nonunique or enantiomeric forms, 9 and 7 stable conformers for 103 and 105, respectively. The three lowest-energy forms for each diastereomer, representing a combined population of >95% (Boltzmann distribution), are listed in Table 28, together with their MM2 calculated energies. Coupling constants between the vicinal protons at C-1 and C-2 in these structures were calculated from the appropriate dihedral angles and compared with the NMR spectra of the two products. The assigned stereochemistry was further supported by additional structural parameters, such as the relative orientation of the C-methyl group with respect to the benzene ring, leading to a different degree of deshielding and consequently different chemical shifts. Based on the MM2 calculated energies (Table 28), a 105:103 ratio of 3.3:1 was calculated, in accord with the experimental findings which suggest higher thermodynamical stability for 105

1. Molecular mechanics calculations |
75 |
TABLE 28. The most stable conformations of trans- and cis-2,3,4,5,6,7-hexahydro-3,7-dimethyl-1- phenyl-1H-3-benzazonine (103 and 105) as calculated by MM2-80
Conformer |
Ga |
% Composition |
Conformer |
Ga |
% Composition |
103a |
24.71 |
67.87 |
105a |
24.08 |
59.76 |
103b |
25.42 |
20.48 |
105b |
24.54 |
27.49 |
103c |
25.99 |
7.82 |
105c |
25.35 |
7.82 |
aIn kcal mol1. A zero change in S was assumed.
(although the ‘real’ thermodynamic ratio could not have been determined experimentally because of the irreversible transformation into the elimination product; see above). A similar analysis was carried out for the 103 precursor, 102. Here an MMP1-based conformational search (The MMP1 program includes the MM1 force field with conjugated-system calculations; at that time, the MM2-80 program did not include this option) located 8 conformers with an overwhelming preference for the most stable one (a Boltzmann population of 83% at 25 °C). In this conformer (102a), the exocyclic methylene is not coplanar with the fused benzene ring (in accord with the UV spectrum), and its top face is thus more exposed to reactants. This simple model, which neglects solvation and complexation effects, still predicts a substantial preference for one diastereomeric product, in partial agreement with the experimental data which suggest complete diastereoselectivity (i.e. only one diastereomer, 103, is formed in the hydrogenation of 102).
N
(102a)
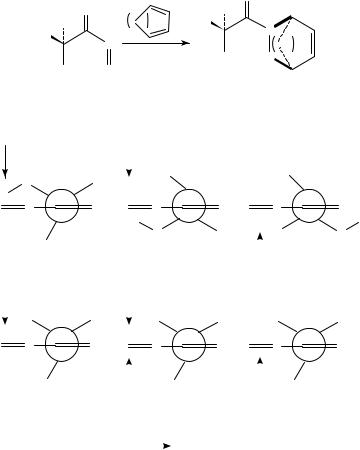
The MM2 force field as implemented in MacroModel 2.0 was used by Miller and Procter in an attempt to rationalize the different diastereoselectivity observed in the cycloaddition of the chiral acyl-nitroso compounds 106a and 106b with cyclopentaand cyclohexadienes (Scheme 9)126. Since the anti arrangement of the acyl-nitroso group was calculated, both by ab initio and molecular mechanics, to be 2.8 kcal mol 1 more stable than the syn one, only the former needed to be considered. The calculated, three lowest rotamers of 106a and 106b about the N(DO) C(DO) C O torsion are shown in Scheme 10, together with their MM2 relative energies and the preferred directions for a diene attack, based on the relative steric hindrance at the two faces of the nitroso bond. For the hydroxy derivative (106a) both the lowest and second-lowest energy conformers exhibit a preference for the same direction of a diene attack. A similar preference is also deduced for the global
76 |
Pinchas Aped and Hanoch Senderowitz |
|
||
|
|
|
O |
|
|
O |
H |
|
|
H |
n |
RO |
N |
|
RO |
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
n = 1,2 |
Ph |
O |
n |
|
|
|
||
Ph |
O |
|
|
|
(106)(a) R= H
(b) R= Me
SCHEME 9
|
|
O |
H |
|
|
|
H |
|
Ph |
|
|
H |
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
||
O |
N |
O |
O |
|
N |
|
O |
O |
N |
O |
|
|
|
|
|
|
H |
|
|
|
|
H |
|
|
|
Ph |
|
|
|
O |
|
Ph |
|
H |
O |
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
0.00 |
|
|
0.70 |
|
|
|
0.88 |
||
|
|
|
|
|
|
|
|||||
|
|
H |
Ph |
|
MeO |
|
H |
|
Ph |
OMe |
|
|
|
|
|
|
|||||||
O |
N |
O |
O |
|
N |
|
O |
O |
N |
O |
|
|
|
MeO |
|
|
|
|
Ph |
|
|
H |
|
|
|
|
|
|
|
|
|
||||
|
|
0.00 |
|
|
|
|
0.61 |
|
|
0.86 |
|
|
|
|
|
|
|
|
|
= attack of diene |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
SCHEME 10. The three lowest-energy rotamers of the acyl-nitroso compounds, 106a (upper row) and 106b (lower row), as calculated by MM2. Relative energies are in kcal mol 1
minimum of the methoxy derivative (106b). However, for the second lowest energy form of this system, the diene approach is almost equally hindered from both sides of the NDO bond. Indeed, cycloadditions involving the methoxy derivative are experimentally known to be less selective than those involving the hydroxy one.
D. Heat of Formation and Density Calculations of Energetic Materials
Among the most important characteristics of energetic materials are their heat of formation and density. The former is a measure of the energy capacity that is released during explosion, while the latter is directly related to performance parameters such as detonation velocity and pressure. The experimental difficulties and hazards in treating such compounds further incite the development of theoretical methods for predicting their needed properties. Organic nitro and N-nitro derivatives are an important class of energetic materials.

1. Molecular mechanics calculations |
77 |
Akutsu and coworkers127 129 have tried to establish the capability of low-cost computational methods, such as semiempirical molecular orbitals and molecular mechanics calculations, to provide accurate heats of formation for nitro compounds. The recent parameterization of the most popular force fields, MM2 and MM3, as well as Osawa’s version of MM2, MM20, for nitro compounds, has been described in Section II of this chapter. The excellent performance of the first two in calculating heats of formation of such systems is demonstrated in Table 18, showing standard deviations of less than 0.3 kcal mol 1 for both force fields. The main problem is still the very small and limited series of compounds used for parameterization. Thus, the parameter set does not cover cases of poly nitro compounds, where the nitro groups substitute the same, or neighboring, carbons. Similarly, aromatic nitro compounds were not included in the experimental data set and N-nitro derivatives were completely ignored. The parameters used by Akutsu were those determined by Tanabe and collaborators15 for nitro compounds and by Lauderdale and Rodgers130 for nitramines. The results (i.e. gas-phase heats of formation), as calculated by the MM2 and MM20 force fields for a series of aliphatic and aromatic nitro compounds, are presented in Table 29 with their deviations from the experimentally observed values. As evident from the data, the largest errors occur when more
TABLE 29. MM2 and MM20 calculated gas-phase heats of formation for aliphatic and aromatic nitro compounds
Compound |
Experiment |
Referencea |
MM2-Exp.b |
MM20 -Exp.c |
Nitromethane |
17.8 |
P |
C1.3 |
0.5 |
Nitroethane |
24.4 |
P,C |
0.1 |
|
1-Nitropropane |
29.6 |
P |
C0.8 |
C0.7 |
2-Nitropropane |
33.2 |
P,C |
C2.2 |
C3.4 |
1-Nitrobutane |
34.4 |
P,C |
0.2 |
1.8 |
2-Nitrobutane |
39.1 |
P,C |
C2.5 |
C3.6 |
Dinitromethane |
14.1 |
P |
C3.0 |
C1.7 |
1,1-Dinitroethane |
24.1 |
I |
|
C6.3 |
1,2-Dinitroethane |
22.9 |
I |
C1.6 |
2.7 |
1,1-Dinitropropane |
24.1 |
P |
C1.6 |
|
1,3-Dinitropropane |
31.6 |
I |
|
1.1 |
2,2-Dinitropropane |
27.0 |
I |
|
5.4 |
1,1-Dinitrobutane |
34.1 |
I |
|
C5.8 |
1,4-Dinitrobutane |
38.9 |
I |
C8.8 |
1.9 |
Trinitromethane |
0.05 |
P |
C1.5 |
|
1,1,1-Trinitroethane |
12.4 |
I |
|
C11.6 |
1,1,1-Trinitropropane |
18.4 |
I |
C80.1 |
C14.3 |
Tetranitromethane |
19.2 |
C |
C11.2 |
|
Nitrobenzene |
16.1 |
P |
1.2 |
|
o-Nitrotoluene |
9.3 |
K |
0.6 |
|
m-Nitrotoluene |
4.1 |
K |
C2.8 |
|
p-Nitrotoluene |
7.4 |
P |
0.5 |
|
o-Dinitrobenzene |
20.2 |
K |
C2.6 |
|
m-Dinitrobenzene |
12.9 |
P |
0.9 |
|
p-Dinitrobenzene |
13.3 |
K |
2.0 |
|
1,3,5-trinitrobenzene |
14.9 |
P |
4.7 |
|
2,4,6-Trinitrotoluene |
10.4 |
P |
0.4 |
|
aValues in kcal mol 1. When several sources used for the theoretical works gave different experimental values as reference, the latest was quoted; C D J. D. Cox and G. Piltcher, Thermochemistry of Organic and Organometallic Compounds, Academic Press, London, 1969; P D J. B. Pedley, R. D. Naylor and S. P. Kirby, Thermochemical Data of Organic Compounds, 2nd ed., Chapman and Hall, London, 1986; I D B. I. Istomin and V. Palm, Reakt. Sposobnosi Org. Soedin., 10, 583 (1973); K D Kagakukai, Nippon, Kagakubinran, Maruzen, Tokyo, 1984.
bReference 127. cReference 128.

78 |
Pinchas Aped and Hanoch Senderowitz |
than one nitro group reside on the same carbon. When such cases are excluded, the fit between theory and experiment seems quite reasonable with standard deviations of 1.6 and 2.1 kcal mol 1 for the aliphatic series, as calculated by MM2 and MM20, respectively, and 2.2 kcal mol 1 for the aromatic compounds, as calculated by MM20. Next, condensed-phase heats of formation, which are more relevant for energetic compounds, were estimated by combining HV and Hs obtained from additivity rules (the authors used Laidler’s values for HV131 and Bondi’s values for Hs132) with the MM20 calculated gas-phase H0f . Several results are given in Table 30 and are less satisfactory
than the gas-phase numbers, with standard deviations of 3.0 and 5.6 kcal mol 1 for the liquid and solid phases, respectively (these rather large standard deviations were obtained even when excluding the worst discrepancies, such as 2-methyl-2,3,3-trinitrobutane and -propane, which are claimed to have large errors in the experimental values133, and 1,1,1- trinitroethane which already had a very large error in the gas-phase calculated H0f ; see Table 29). These authors have also calculated the heats of formation of N-nitro compounds (nitramines)129, using a value of 11.1 kcal mol 1 for the missing N NO2 bond enthalpy increment. The results (Table 31) are of a comparable quality: 3.9 kcal mol 1 (standard deviation) for the gas-phase heats of formation, and 6.0 kcal mol 1 (after exclusion of the very nitro-crowded 1,1,1,3,5,5,5-heptanitro-3-azapentane) for the solid phase.
Traditional methods to estimate solid-phase density from molecular structure are primarily based on a simple summation of appropriate atomic or group volumes. The basic disadvantage of these ‘group or volume additivity’ procedures134 is that they disregard crystal-packing efficiency and molecular conformation. Thus, conformational isomers, or even different compounds with the same functional group composition, will all be calculated to have the same density. To solve this problem, Ammon and coworkers have developed a scheme to estimate molecular densities by predicting possible crystal
TABLE 30. MM20 calculated condensed-phase heats of formation for aliphatic nitro compounds
|
|
Liquid phase |
|
|
|
Solid phase |
|
Compound |
Experiment |
referencea |
MM20 -Exp.b |
Experiment |
reference |
MM20 -Exp. |
|
1-Nitrobutane |
46.0 |
C |
2.0 |
|
|
|
|
2-Nitrobutane |
49.6 |
C |
C2.4 |
|
|
|
|
Dinitromethane |
25.1 |
C |
2.4 |
|
|
|
|
1,1-Dinitroethane |
34.7 |
S |
C0.8 |
42.0 |
|
5.8 |
|
1,2-Dinitroethane |
40.1 |
|
C0.4 |
C |
|||
1,1-Dinitropropane |
C |
|
|
|
|
||
1,3-Dinitropropane |
53.5 |
C |
C3.6 |
44.9 |
|
0.4 |
|
2,2-Dinitropropane |
|
|
|
C |
|||
2,2-Dimethyl- |
|
|
|
66.4 |
|
9.0 |
|
1,3-Dinitropropane |
|
|
|
S |
|||
1,1,1-Trinitroethane |
28.1 |
|
C0.2 |
27.5 |
S |
C14.7 |
|
1,1,1-Trinitropropane |
S |
|
|
|
|
||
2-Methyl- |
|
|
|
79.2 |
|
C21.6 |
|
2,3,3-trinitrobutane |
|
|
|
C |
|||
2-Methyl- |
|
|
|
69.4 |
|
C6.3 |
|
2,3,3-trinitropentane |
|
|
|
C |
|||
1,1,1,4-Tetranitrobutane |
42.8 |
P |
3.1 |
45.3 |
P |
C6.0 |
|
2,2,3,3-Tetranitrobutane |
40.1 |
P |
C6.6 |
43.5 |
P |
1.8 |
|
aValues in kcal mol 1. For references, see Tables 29; S D D. R. Stull, E. F. Westrum Jr. and G. E. Sinke, The Chemical Thermodynamics of Organic Compounds, Wiley, New York, 1969.
bReference 128.

|
1. Molecular mechanics calculations |
|
79 |
||||
TABLE 31. MM20 calculated gasand solid-phase heats of formation for several nitramines |
|||||||
|
|
Gas phase |
|
|
|
Solid phase |
|
|
|
|
|
|
|
|
|
Compound |
experiment |
referencea |
MM20 -Exp.b |
experiment |
reference MM20 -Exp. |
||
DMN (107) |
0.2 |
C |
C1.1 |
16.9 |
C |
C4.1 |
|
DEN |
12.7 |
P |
C2.0 |
12.7 |
|
C7.3 |
|
1,4-Dinitropiperazine |
13.9 |
P |
C7.2 |
P |
|||
RDX (108) |
46.8 |
U |
4.0 |
14.7 |
S |
4.0 |
|
HMX (109) |
56.4 |
U |
C1.4 |
17.9 |
S |
2.9 |
|
DNNC (110) |
|
|
|
2.0 |
B |
6.3 |
|
2,2,4,6,6-Pentanitro- |
|
|
|
55.4 |
|
6.9 |
|
4-azaheptane |
|
|
|
P |
|||
1,3,3,5,7,7-Hexanitro- |
|
|
|
5.0 |
|
C8.4 |
|
1,5-diazaoctane |
|
|
|
B |
|||
1,1,1,3,5,5,5-Heptanitro- |
|
|
|
6.7 |
|
C14.3 |
|
3-azapentane |
|
|
|
P |
|||
aValues in kcal mol 1. For references, see Tables 29 and 30; B D Reference 133. bReference 129.
|
|
|
|
|
NO2 |
|
|
|
NO2 |
|
|
|
|
|
|
N |
|
O2 N |
N |
||
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
N |
N |
|
N |
||||
(CH3 )2 N |
|
NO2 |
O2 N |
|
NO2 |
N |
NO2 |
|||
|
||||||||||
|
|
|
|
|
|
|
|
|
O2 N |
|
(107) |
|
(108) |
|
|
|
(109) |
||||
|
|
NO2 |
|
|
|
|
||||
O2 N |
|
|
|
|
|
|
NO2 |
NO2 |
||
|
|
N |
|
NO2 |
|
|
||||
|
|
|
|
|
|
|
||||
O2 N |
|
|
|
|
|
|
|
|
||
|
N |
(CH3 )2 C |
|
|
C(CH3 )2 |
|
|
|||
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
||
|
NO2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NO2 |
|
|
|||
(110) |
(111) |
(112) |
|
|||||||
|
|
|
NO2 |
|
H3 C |
C6 H6 |
|
|||
|
|
|
|
|
NH |
|
||||
O2 N |
|
|
|
|
|
|
|
|||
N |
|
|
|
|
||||||
|
|
|
|
|
|
|
||||
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|||
|
|
N |
|
|
NO2 |
|
O2 N |
NO2 |
||
|
|
|
|
|
|
|||||
|
|
NO2 |
|
|
|
|
|
|
|
|
(113) |
|
|
|
|
|
|
(114) |
|
||

80 |
Pinchas Aped and Hanoch Senderowitz |
structures, and incorporated it into their MOLPAK computer program135. The procedure operates according to the following steps: (1) Obtaining the geometry of the desired molecule in isolation (usually, but not always, of the most stable conformer) either by calculation, or by taking a known structure from a data base. The preferred source in MOLPAK is a semiempirical AM1 calculated geometry. (2) Obtaining the crystallographic symmetry, i.e. the definition of the unit cell and the crystallographic space group. This, in principle, can be obtained from an external source, such as the Cambridge Structural Database (CSD). In MOLPAK, a set of group symmetries is calculated, using rules stored in subgroups or ‘patterns’ relating to the various space groups (the January 92 version of MOLPAK covers crystal types belonging to ca 83% of the structures found in the CSD).
(3) Defining the size of the 3D model of the crystal lattice and building it by applying the appropriate symmetry rules. The size of the model is taken as the ‘coordination sphere’ of the molecule which includes all neighboring molecules in a close VdW contact with it. An analysis of experimentally determined structures gave an average number of fourteen molecules in the ‘coordination sphere’. (4) Each initial lattice model is then refined, without changing the internal molecular geometry (rigid-body approximation), using a central force field (WMIN) of the general form given in equation 63:
n |
N |
|
Wc D 1/2Z |
qiqj/rij AiAj/rij6 C BiBj exp[ Ci C Cj rij] |
63 |
i |
i6Dj |
|
where Z is the number of molecules per unit cell, Wc is the lattice energy (in kcal mol 1), A, B and C are energy coefficients, qi are atomic charges, rij are interatomic distances, and n and N are the number of atoms in one, and all, unit cells, respectively.
To overcome the problem of obtaining accurate values for the atomic charges, the Coulombic term in equation 63 was eliminated and replaced by an appropriate parameterization of A, B and C to fit a set of known structures. Other schemes, such as the PCK/ME procedure of Ritchie136 which uses a model of atom-centered multipoles (ACME), may also be used at this stage. (5) The dimensions of the unit cell are calculated from the refined lattice, and used to determine its volume and density. The entire process is iterative, and provides a set of possible crystal structures for the studied molecule. The energy coefficients, A, B and C, of the WMIN force field have been determined for the first-row elements C, H, N, O and F, and their values are given in Reference 135. A series of nitro and nitramine compounds were studied by the MOLPAK program. Selected results, presented as molecular volumes, are given in Table 32, and show a very good fit (SD D 1.08%) with the experimental data.
TABLE 32. Unit cell volume per molecule (A˚ 3) as calculated by MOLPAK and observed experimentally, for several nitro compounds and nitramines
|
|
V/Z |
Compound |
calculated |
observeda |
111 |
411.15 |
409.20 |
112 |
233.31 |
229.43 |
113 |
590.99 |
597.43 |
114 |
346.50 |
344.32 |
aTaken from the Cambridge Structural Database (CSD).