

Раздел 5.2
Наиболее распространённые энзимопатии.
Врождённые нарушения обмена фенилаланина и тирозина
На рисунке 5.2 представлена схема основных химических превращений фенилаланина и тирозина, а также известных в настоящее время нарушений активности ферментов, катализирующих эти реакции. Из схемы видно, что тирозин, который в норме образуется в организме из фенилаланина, является предшественником целого ряда биологически активных соединений.
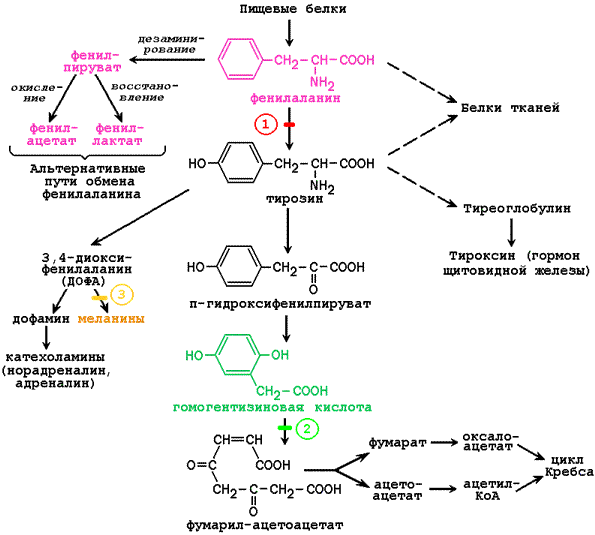
Рисунок 5.2. Обмен фенилаланина и тирозина и возможные нарушения. Цифрами показаны блокированные реакции при следующих заболеваниях: 1 - фенилкетонурия; 2 - алкаптонурия; 3 - альбинизм.
Фенилкетонурия. Это заболевание вызывается дефицитом печёночного фермента фенилаланингидроксилазы, или реже, нарушением биосинтеза его кофактора тетрагидробиоптерина. Поскольку фенилаланингидроксилаза катализирует превращение фенилаланина в тирозин, при фенилкетонурии в крови накапливается фенилаланин, который наряду с продуктами его альтернативных превращений (фенилпируват, фениллактат, фенилацетат) экскретируется с мочой. Присутствие в моче пациентов фенилпирувата (соединения, содержащего кетогруппу) нашло отражение в названии заболевания.
Избыток фенилаланина вызывает замедление транспорта тирозина и других аминокислот через клеточные мембраны. Следствием этого может быть нарушение обмена аминокислот в клетках головного мозга с последующим расстройством биосинтеза белка и нарушением синтеза нейромедиаторов. Если своевременно не выявить дефект фермента и не начать лечение, в течение первого года жизни у детей развивается умственная отсталость. В связи с этим в ряде стран широко практикуются скрининговые исследования новорождённых с целью раннего выявления случаев фенилкетонурии (определение концентрации фенилаланина в крови, взятой из прокола кожи на пятке).
Лечение заболевания заключается в том, чтобы ограничить поступление фенилаланина с пищей. С этой целью используются искусственные питательные смеси. По современным рекомендациям, длительность лечения фенилкетонурии составляет от 5 до 10 лет.
Алкаптонурия. Это заболевание обусловлено врождённой недостаточностью оксидазы гомогентизиновой кислоты. Гомогентизиновая кислота накапливается в крови, тканях и моче. Окисление и полимеризация этого вещества приводит к образованию пигмента алкаптона. Отложение алкаптона в хрящах, которые затем темнеют, называется охронозом. Превращению гомогентизиновой кислоты в алкаптон способствует щелочная среда; при алкаптонурии наиболее явным симптомом является экскреция либо чёрной мочи, либо мочи, которая темнеет по мере защелачивания при хранении.
Алкаптонурия в большинстве случаев не требует специального лечения, но в среднем возрасте и позже обычно развивается артрит.
Альбинизм. Недостаточность фермента тирозиназы в меланоцитах (пигментных клетках кожи и радужной оболочки глаз) вызывает одну из форм альбинизма и наследуется как рецессивный признак. У пациентов отсутствует пигментация кожи, волос и радужной оболочки (глаза кажутся розовыми). Отсутствие пигмента в коже сопровождается повышенной чувствительностью к солнечным лучам. Следует отметить, что биосинтез адреналина у альбиносов не нарушается, так как тирозиназа, участвующая в образовании катехоламинов, представляет собой другой фермент, контролируемый иным геном.
Галактоземия
Галактоземия - врождённое нарушение обмена веществ, обусловленное недостаточностью фермента галактозо-1-фосфатуридилтрансферазы. Для галактоземии характерна триада симптомов: увеличение размера печени, катаракта и умственная отсталость. В крови больных повышено содержание галактозы, этот моносахарид обнаруживается и в моче. Первые признаки заболевания у ребёнка (диарея, рвота, обезвоживание) выявляются очень рано, обычно через несколько дней после начала грудного вскармливания.
Источником галактозы в организме является дисахарид лактоза, содержащаяся в молоке. После расщепления лактозы в микроворсинках слизистой оболочки тонкого кишечника галактоза под действием фермента галактокиназы превращается в печени в галактозо-1-фосфат (рисунок 5.3).
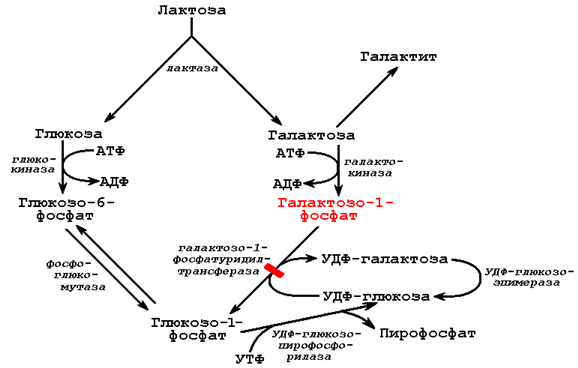
Рисунок 5.3. Обмен галактозы и основная причина галактоземии.
В нормальных условиях галактозо-1-фосфат под влиянием галактозо-1-фосфатуридилтрансферазы переходит в УДФ-галактозу. В результате угнетения этой реакции в организме накапливается галактозо-1-фосфат - метаболит с очень коротким в нормальных условиях периодом существования. В связи с этим в норме он не вызывает нарушений. Однако при накоплении галактозо-1-фосфата проявляется его мощное токсическое действие. Природа токсического влияния галактозо-1-фосфата, вероятно, объясняется структурным сходством галактозы с глюкозой. Галактозо-1-фосфат, присоединяясь к активному центру ферментов, метаболизирующих глюкозо-1-фосфат, блокирует их, что приводит к нарушению обмена глюкозы. Так, в клетках печени накопление галактозо-1-фосфата вызывает ингибирование фосфоглюкомутазы и глюкозо-6-фосфатазы - ферментов, участвующих в превращении гликогена в глюкозу, в результате чего снижается уровень глюкозы в крови. В хрусталике глаза избыток галактозы переходит в шестиатомный спирт галактит. Галактит не подвергается дальнейшим превращениям и приводит к набуханию соединительной ткани и развитию катаракты. В клетках головного мозга нарушается синтез гликолипидов вследствие недостаточного образования их предшественника УДФ-галактозы.
Если галактозу не исключить из диеты, возможны тяжёлые последствия, в том числе летальный исход.
Гликогенозы
Этот термин является общим для группы наследственных заболеваний, характеризующихся отложением в тканях аномально больших количеств полисахарида - гликогена, являющегося важным источником энергии и резервом углеводов в тканях. Врождённые нарушения содержания и структуры гликогена обусловлены дефицитом одного из ферментов, участвующих в расщеплении гликогена в печени или в скелетных мышцах (рисунок 5.4).
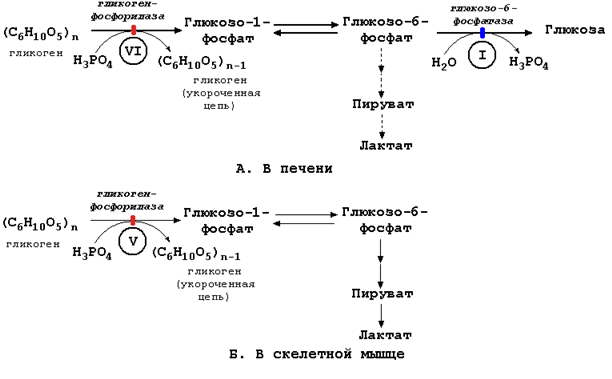
Рисунок 5.4. Расщепление гликогена в печени и скелетных мышцах и его нарушения.
Примеры:
Гликогеноз I типа (болезнь Гирке) – дефицит глюкозо-6-фосфатазы в печени. Характеризуется повышенным содержанием гликогена в печени; содержание глюкозы в крови снижено. После введения адреналина или глюкагона (гормонов, активирующих фермент гликогенфосфорилазу), уровень пирувата и лактата в крови существенно возрастает.
Гликогеноз V типа (болезнь Мак-Ардля) – дефицит фосфорилазы в скелетных мышцах. У больных развивается пониженная выносливость к физическим нагрузкам. В скелетных мышцах содержится аномально высокое количество гликогена. Тем не менее, после выполнения физической работы или после введения адреналина содержание лактата в крови не увеличивается.
Гликогеноз VI типа (болезнь Херса) – дефицит фосфорилазы в печени. Для этого заболевания характерно повышение содержания гликогена в печени, гипогликемия. После введения адреналина или глюкагона содержание лактата в крови не увеличивается (в отличие от гликогеноза I типа).