

Baer M., Billing G.D. (eds.) - The role of degenerate states in chemistry (Adv.Chem.Phys. special issue, Wiley, 2002)
.pdf302 aron kuppermann and ravinder abrol
section as wð1ÞadðRlÞ. For a triatomic system, this vector is six dimensional (6D).
As discussed in Section II.A, the adiabatic electronic wave functions celi ;ad and celj ;ad depend on the nuclear coordinates Rl only through the subset ql (which in the triatomic case consists of a nuclear coordinate hyperradius r and a set of two internal hyperangles nl), this permits one to relate the 6D vector wð1ÞadðRlÞ to another one wð1ÞadðqlÞ that is 3D. For a triatomic system, let aIl ðaIl; bIl; cIlÞ be the Euler angles that rotate the space-fixed Cartesian
frame into the body-fixed principal axis of inertia frame Il, and let $Il be the
Rl
6D gradient vector in this rotated frame. The relation between the space-fixed
$R |
and $Il |
is given by |
|
|
|
l |
R |
|
|
|
|
|
l |
|
|
|
|
|
|
~ |
Il |
Il |
ð60Þ |
|
|
$Rl ¼ Rða |
|
Þ $Rl |
where RðaIlÞ is a 6 6 block-diagonal matrix whose two diagonal blocks are
both equal to the 3 3 rotational matrix RðaIlÞ. The $Il operator can be
Rl
written as [83]
Il |
|
Il |
ðnlÞp |
Il |
|
Il |
|
^Il |
ða |
Il |
Þ |
|
|
ð61Þ |
$Rl ¼ G |
|
|
ðqlÞ þ H |
ðnlÞJ |
|
|
|
|||||||
|
|
|
^ |
|
|
|
|
|
|
|
|
|
|
|
In this expression, GIl |
and |
HIl are |
both 6 3 |
rectangular matrices |
whose |
|||||||||
elements are known functions of the internal hyperangles nl. |
^Il |
is a 3 1 |
||||||||||||
p |
||||||||||||||
column vector operator whose elements contain first derivatives with respect to
the three |
ql coordinates and |
^Il |
is the 3 |
1 column vector operator whose |
|||
J |
|
||||||
|
|
^Il |
|
^Il |
|
^Il |
of the system’s nuclear motion |
elements are the components Jx |
, Jy |
, and Jz |
|||||
|
|
|
|
|
^ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
angular momentum operator J in the Il frame. From these properties, it can be |
|||||||||||||||||||||||||||||
shown that |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
wð1ÞadðRlÞ ¼ RðaIlÞGIlðnlÞwð1ÞadðqlÞ |
|
|
|
|
|
ð62Þ |
||||||||||||||||||||
and that |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Wð1Þad |
ð |
R |
lÞ $Rl v |
ad |
ð |
R |
|
|
GIl |
ðxlÞ |
Wð1Þad |
ð |
q |
l |
|
Il |
ad |
ð |
R |
lÞ |
ð |
63 |
Þ |
||||||
|
|
|
lÞ ¼ |
|
|
|
|
|
|
Þ $Rl v |
|
|
|
||||||||||||||||
where |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
wð1Þad |
q |
|
|
el;ad |
ð |
r; q |
lÞj |
p^Il |
ð |
q |
|
el;ad |
ð |
r; q |
lÞir |
|
|
|
ð |
64 |
Þ |
||||||||
|
|
|
ð |
lÞ ¼ hc1 |
|
|
|
|
lÞc2 |
|
|
|
|
|
|
|
|||||||||||||
is a 3D column vector and Wð1ÞadðqlÞ is a 2 2 skew-symmetric matrix whose only nonzero element is the wð1ÞadðqlÞ vector. By using the symmetrized hyperspherical coordinates defined in Section IV.A for a triatomic system, the
quantum reaction dynamics for multiple electronic states 303
elements of p^Il are the spherical polar components of the 3D gradient associated with the polar coordinates r; y; fl [84]:
|
|
|
|
|
0 |
|
q |
|
1 |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
qr |
|
|
|
|
|
|
|
||||||
|
|
|
p^Il |
¼ |
1 |
q |
|
|
|
|
|
ð |
65 |
Þ |
|||||
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
B |
1r qy q |
C |
|
|
|
|
|
||||||||
|
|
|
|
|
@ |
|
|
|
|
|
|
A |
|
|
|
|
|
|
|
|
|
|
|
|
B |
r sin y |
|
qfl |
C |
|
|
|
|
|
|
|
|||
The corresponding cartesian gradient $ql is given by |
|
|
|
|
|
|
|
||||||||||||
$ql |
¼ |
|
siny cosfl |
|
cosy cosfl |
sinfl |
1p^ |
I |
|
ð66Þ |
|||||||||
0 siny sinfl |
|
cosy sinfl |
cosfl |
|
l |
||||||||||||||
|
|
@ |
cosy |
|
|
siny |
|
0 |
A |
|
|
|
|
|
|||||
in a space whose polar coordinates are r; y; fl.
The wð1ÞadðqlÞ vector can also be decomposed into a longitudinal and a transverse part
wð1ÞadðqlÞ ¼ $ql aðqlÞ þ wtrað1ÞadðqlÞ |
ð67Þ |
where, aðqlÞ is a scalar potential. It can be shown using Eq. (58) and the two- electronic-state counterpart of Eq. (47) that bðqlÞ ¼ aðqlÞ. The diabatization angle bðqlÞ can be obtained by taking the divergence of Eq. (67) and solving for the resulting Poisson equation
rq2l bðqlÞ ¼ sðqlÞ |
ð68Þ |
where
sðqlÞ ¼ rql wð1ÞadðqlÞ |
ð69Þ |
is known because wð1ÞadðqlÞ has been accurately calculated and fitted over the entire ql space of interest [84]. The nuclear–electronic rotational couplings associated with the rotation of the H3 molecular plane relative to a space-fixed frame vanish identically if the mass-scaled nuclear and electronic coordinates of Eqs. (4) and (5) are used and the electronically adiabatic PESs are calculated accordingly. This is not, however, done in standard electronic structure calculations, as mentioned after Eq. (8), and as a result such couplings do not vanish. We have, however, in our electronic wave function calculations [84], found them to be at least two orders of magnitude smaller than the jwð1ÞadðqlÞj.
304 |
aron kuppermann and ravinder abrol |
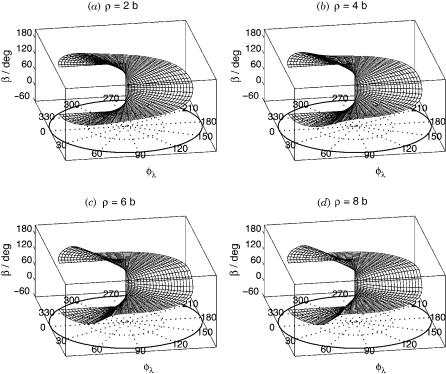
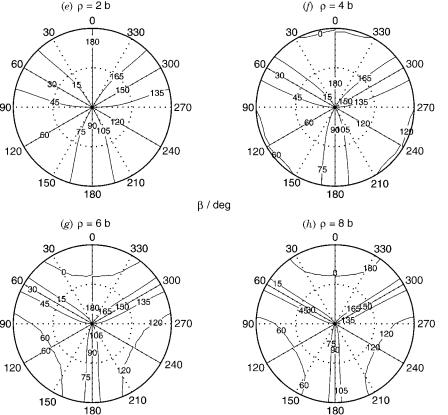
Figure 2. The diabatization angle bðr; y; flÞ, in degrees, for the H3 system at (a) r ¼ 2 b, (b) r ¼ 4 b, (c) r ¼ 6 b, and (d) r ¼ 8 b. The equatorial view of b contours is also given at (e) r ¼ 2 b, (f) r ¼ 4 b, (g) r ¼ 6 b, and (h) r ¼ 8 b.
This justifies the use of the simpler ql language over the Rl one. The solution of the Poisson equation and the boundary conditions used are explained in detail elsewhere [55]. Here, we will present some selected results.
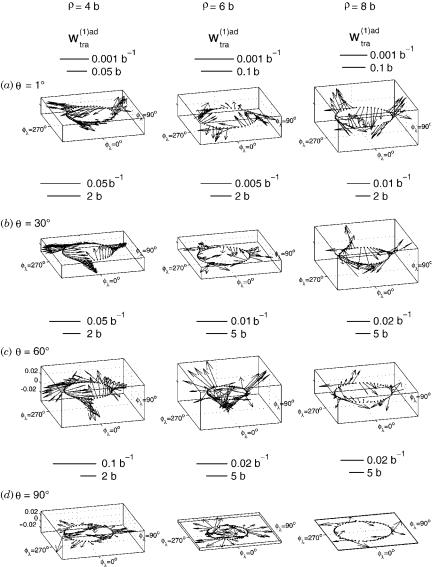
The internal coordinates used in the calculation are the hyperradius r, and the hyperangles y and fl, described in Section IV.A. The equation y ¼ 0 corresponds to conical intersection geometries and y ¼ 90 to collinear ones. For a fixed r and y, as fl is varied from 0 to 2p, the system executes a loop in internal configuration space around the corresponding conical intersection geometry. In Figure 2, the diabatization angle b is displayed for several values of r as a function of y and fl. Use of this b and Eq. (67), furnishes the transverse part wðtra1ÞadðqlÞ over the entire dynamically important region of internal configuration space. This is displayed in Figure 3. These sets of b and wðtra1ÞadðqlÞ were obtained using an optimal mixture of Dirichlet and Neumann
quantum reaction dynamics for multiple electronic states 305
Figure 2 (Continued)
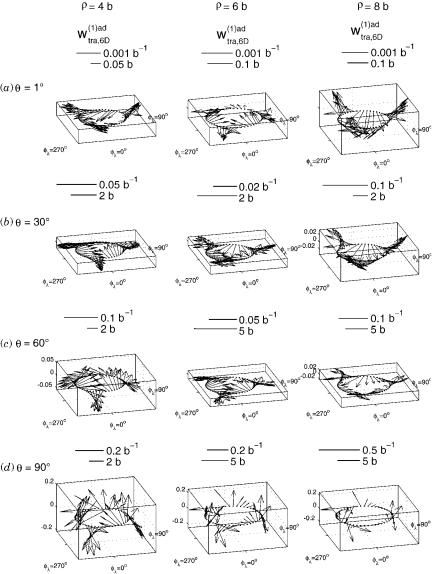
conditions for the solution of the above-mentioned Poisson equation. Using pure Dirichlet conditions instead gives a different transverse part, displayed in Figure 4. Comparison with Figure 3 clearly shows that the optimal boundary conditions significantly reduce the magnitude of the transverse part as compared to all-Dirichlet condition. Comparison of the average magnitude of the transverse vector over the entire internal configuration space for both the optimal and the all-Dirichlet boundary conditions, shows that the optimal condition average was 4.7 times smaller than the all-Dirichlet one. This result indicates that use of the optimal mixed set of Neumann and Dirichlet boundary conditions for solving the Poisson Eq. (68) does indeed significantly reduce the average magnitude of the transverse part of the first-derivative coupling vector.
306 |
aron kuppermann and ravinder abrol |
Figure 3. Transverse (nonremovable) part of the ab initio first-derivative coupling vector, wðtra1Þadðr; y; flÞ as a function of fl for r ¼ 4, 6, and 8 b and (a) y ¼ 1 (near-conical intersection geometries), (b) y ¼ 30 , (c) y ¼ 60 , and (d) y ¼ 90 (collinear geometries).
quantum reaction dynamics for multiple electronic states 307
Figure 4. Same as Figure 3 for transverse (nonremovable) part of the ab initio first-derivative coupling vector wðtra1Þ;ad6Dðr; y; flÞ, obtained using the all-Dirichlet boundary conditions.
308 |
aron kuppermann and ravinder abrol |
|
|||||||||
The vector |
wð1Þad q |
lÞ |
[or |
wð1Þad |
q |
lÞ |
] can |
also |
provide |
a good first |
|
|
ð |
|
1;2 |
|
ð |
|
|
2 ad |
ðqlÞ, which in |
||
approximation to the second-derivative coupling matrix Wð Þ |
|||||||||||
a two-electronic-state approximation is given by |
|
|
|
||||||||
|
Wð2ÞadðqlÞ ¼ |
w1ð21Þad |
|
q |
|
|
w1ð22Þad q |
|
|||
|
wð2;Þad |
ðqlÞ |
|
wð2;Þad |
ðqlÞ ! |
ð70Þ |
|||||
|
|
|
|
2;1 |
ð lÞ |
|
2;2 |
ð lÞ |
|
||
In the two-electronic-state Born–Huang expansion, the full-Hilbert space of adiabatic electronic states is approximated by the lowest two states and furnishes for the corresponding electronic wave functions the approximate closure relation
jc1el;adðr; qlÞihc1el;adðr; qlÞj þ jc2el;adðr; qlÞihc2el;adðr; qlÞj 1 |
ð71Þ |
By using this equation and the fact that for real electronic wave functions the diagonal elements of Wð1ÞadðqlÞ vanish, it can be shown that
wð12;1ÞadðqlÞ ¼ wð22;2ÞadðqlÞ ¼ wð11;2ÞadðqlÞ wð11;2ÞadðqlÞ
wð12;2ÞadðqlÞ ¼ wð22;1ÞadðqlÞ ¼ 0
ð72Þ
For the H3 system, since wð11;2ÞadðqlÞ is known over the entire ql space [84], Eq. (72) can be used to obtain the two equal nonzero diagonal elements of the Wð2ÞadðqlÞ matrix. Since this matrix appears with a multiplicative factor of
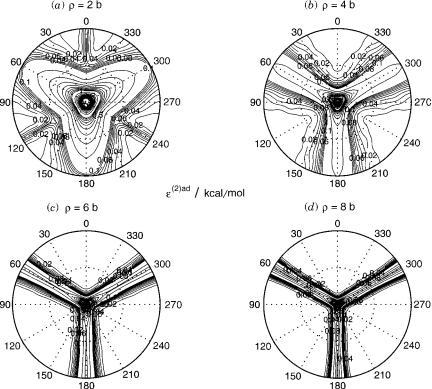
ð h2=2mÞ in the adiabatic nuclear motion Schro¨dinger equation giving it the units of energy, both ð h2=2mÞ wð12;1ÞadðqlÞ and ð h2=2mÞ wð22;2ÞadðqlÞ can be labeled as eð2Þad. In Figure 5, this quantity is displayed in units of kilocalories
per mole (kcal/mol) for several values of r as a function of y and fl. It shows the singular behavior of the diagonal elements of Wð2ÞadðqlÞ at conical intersection geometries (y ¼ 0 ). Being a repulsive correction to the adiabatic energies, this singular behavior prevents any hopping of the nuclei from one electronic state to another in the close vicinity of the conical intersection.
By using the diabatic version of the closure relation (71), and Eq. (59), the elements of the diabatic second-derivative coupling matrix Wð2ÞdðqlÞ of Eq. (36) can be expressed as
wð12;1ÞdðqlÞ ¼ wð22;2ÞdðqlÞ ¼ wð11;2Þad;traðqlÞ wð11;2Þad;traðqlÞ
wð12;2ÞdðqlÞ ¼ wð22;1ÞdðqlÞ ¼ 0
ð73Þ
quantum reaction dynamics for multiple electronic states 309
Figure 5. Second-derivative coupling term eð2ÞadðqlÞ defined at the end of Section III.D for the H3 system at (a) r ¼ 2 b, (b) r ¼ 4 b, (c) r ¼ 6 b, and (d) r ¼ 8 b. The following contours are displayed 0–0.1 kcal/mol every 0.01 kcal/mol, 0.1–1.0 kcal/mol every 0.1 kcal/mol and 1.0–10.0 kcal/mol every 0.5 kcal/mol.
where, both ð h2=2mÞ wð12;1ÞdðqlÞ and ð h2=2mÞ wð22;2ÞdðqlÞ can be labeled as eð2ÞdðqlÞ. The values of this (approximate) eð2ÞdðqlÞ calculated from this
equation are smaller than 0.08 kcal/mol over the entire nuclear configuration space involved, and to a very good approximation can be neglected.
IV. TWO-ELECTRONIC-STATE QUANTUM REACTION DYNAMICS
FORMALISM FOR TRIATOMIC REACTIONS
In a two-lowest-electronic-state Born–Huang description for a chemical reaction, the nuclei can move on both of two corresponding PESs during the reaction, due to the electronically non-adiabatic couplings between those states. A reactive scattering formalism for such a reaction involving a triatomic system
310 |
aron kuppermann and ravinder abrol |
is presented below. This formalism is an extension of the time-independent coupled-channel hyperspherical method [26,33–37] that has been used in the past to study triatomic reactions on a single adiabatic electronic state.
A.Symmetrized Hyperspherical Coordinates
Consider a triatomic system with the three nuclei labeled Aa, Ab, and Ag. Let the arrangement channel Al þ AnAk be called the l arrangement channel, where lnk is a cyclic permutation of abg. Let R0l; r0l be the Jacobi vectors associated with this arrangement channel, where r0l is the vector from An to Ak and R0l the vector from the center of mass of AnAk to Al. Let Rl; rl be the corresponding mass-scaled Jacobi coordinates defined by
Rl ¼ |
ml;nk |
|
1=2 |
|
rl ¼ |
mnk |
|
1=2 |
|
|
Rl0 |
and |
rl0 |
ð74Þ |
|||||||
m |
m |
where mnk is the reduced mass of AnAk, ml;nk the reduced mass of the Al; AnAk pair, and m the system’s overall reduced mass given by
m ¼ |
|
m mbmg |
|
|
1=2 |
|
|
|
|
||||
|
a |
|
|
|
||
ma |
mb |
þ |
mg |
|
||
|
|
þ |
|
|
|
|
ml being the mass of atom Al (l ¼ a; b; g). We define a set of symmetrized hyperspherical coordinates r; ol; gl [85,86] by
r ¼ Rl2 þ rl2 1=2 |
ð75Þ |
and |
|
Rl ¼ r cosðol=2Þ rl ¼ r sinðol=2Þ 0 ol p |
ð76Þ |
where r is independent of the arrangement channel [60,61]. The corresponding internal configuration space Cartesian coordinates are defined by
Xl ¼ r sinol cos gl |
|
Y ¼ r sinol singl |
ð77Þ |
Zl ¼ r cosol |
|
where gl is the angle between Rl and rl (or R0l and r0l) in the 0 to p range and ol; gl are the polar angles of a point in this space. The alternate internal configuration space symmetrized hyperspherical coordinates y; fl are defined

|
quantum reaction dynamics for multiple electronic states 311 |
||
as |
the polar angles associated with the interchanged |
|
|
axes OXl ¼ OZl, |
|||
|
|
for which |
|
OYl ¼ OXl, and OZl ¼ OYl |
|
||
|
|
¼ Zl ¼ r siny cosfl |
|
|
Xl |
|
|
|
|
¼ Xl ¼ r siny sinfl |
ð78Þ |
|
Yl |
||
|
|
¼ Y ¼ r cosy |
|
|
Z |
|
|
The coordinates r, y and fl are limited to the ranges |
|
|
0 r < 1 0 y p=2 |
0 fl < 2p |
ð79Þ |
The relation between y; fl and ol; gl is [using Eqs. (77) and (78)] |
|
|
siny cosfl ¼ cosol |
|
|
siny sinfl ¼ sinol cosgl |
ð80Þ |
|
cosy ¼ sinol singl |
|
|
Let GxIlyI zIl be a body-fixed frame Il, whose axes are the principal axes of inertia of the three nuclei and whose Euler angles with respect to the space-fixed frame Gxsf ysf zsf are al; bl; cl with G being the center of mass of the three nuclei. The senses of these axes are chosen to result in a 1:1 correspondence between r; y; fl; al; bl; cl coordinates and the space-fixed Cartesian coordinates of Rl and rl. In addition, the Il axes are labeled so as to order the corresponding principal moments of inertia according to
Izl Ixl Iy |
ð81Þ |
Furthermore, let l refer collectively to the five hyperangles ðy; fl; al; bl; clÞ, ql to the three internal coordinates ðr; y; flÞ and Rl to all six hyperspherical coordinates.
The coordinates r; l are called the principal axes of inertia symmetrized hyperspherical coordinates. The nuclear kinetic energy operator in these coordinates is given by
|
|
|
h |
2 |
|
|
|
|
|
|
|
|
|
|
^2 |
ð lÞ |
|
|
|
T^ |
R |
|
|
|
2 |
|
|
T^ |
|
|
|
|
82 |
|
|||||
lÞ ¼ |
2m rRl ¼ |
|
|
2mr2 |
ð |
Þ |
|||||||||||||
nuð |
|
|
rðrÞ þ |
|
|||||||||||||||
^ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
where, TrðrÞ is the hyperradial kinetic energy operator |
|
|
|
||||||||||||||||
|
|
^ |
|
|
|
|
h2 1 q |
5 |
q |
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
TrðrÞ ¼ |
|
2m r5 qr r |
|
qr |
|
ð83Þ |
|||||||||||