

Flow Cytometry - First Principles (Second Edition)
.pdf
|
Leukocytes, Surface Proteins, and the Strategy of Gating |
109 |
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 6.14. The use of CD45 to exclude erythrocytes, platelets, and debris from a lymphocyte gate. The distinction between monocytes and lymphocytes can be ambiguous with either the FSC/SSC or the CD45/SSC gate. Data ®le provided by Marc Langweiler.
that many benchtop ¯ow cytometers have ``parameters to spare,'' current clinical protocols for lymphocyte analysis suggest that antibodies against CD45 be added to all tubes as a third color to be used for gating. By gating on CD45 positivity along with SSC, the gated cells (Fig. 6.14) will not include high numbers of erythrocytes, debris, or platelets (all CD45 negative). In this way gating can be re®ned if you are using a ¯ow cytometer with extra ¯uorescence parameters. The distinction between lymphocytes and monocytes is not, however, helped by this procedure (and the CD45/SSC gate in Fig. 6.14 indicates ambiguity here). Figure 6.15 illustrates CD45/SSC gating in a bone marrow sample, where this type of analysis has proved particularly useful in identifying clusters that are related to various mature and immature hematological lineages.
In fact, this CD45/SSC gating marks the general trend away from using scatter parameters toward a quite di¨erent strategy for ¯ow analysis. The basic problem, as seen above, is that lymphocytes (or any other taxonomic group) are not a homogeneous collection of cells with perfectly delineated physical characteristics; they are mainly homogeneous, but they usually contain at least some cells at the fringes with marginal characteristics. Any gating based on FSC/SSC forces us either to exclude these fringe cells or to include many extraneous cells. With the availability of multicolor instrumentation

110 |
|
Flow Cytometry |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 6.15. Bone marrow from a normal donor showing CD45/SSC clusters. CD45 expression varies as cells mature. Lymph ˆ lymphocytes; Mono ˆ monocytes; L-Blast ˆ lymphoblasts; M-Blast ˆ myeloblasts; Myeloid ˆ neutrophils; and Erythroid ˆ cells of the erythroid lineage. From Loken and Wells (2000).
and multicolor stains, it has become possible to avoid making any of these di½cult gating decisions and to include all cells in the analysis by using stain itself either to gate in or to gate out the cells of interest.
For example, if one is studying the prevalence of a particular subpopulation among lymphocytes, one could stain every peripheral blood mononuclear cell sample with a PE stain for monocytes in addition to an FITC conjugate of the marker of interest. Then, in the analysis stage, one could simply gate out (exclude) from analysis any PE-positive particles and analyze all the PE-negative particles for the percentage that are FITC-positive. Figure 6.16 shows an example of this protocol. By the use of three-color analysis, there are even greater possibilities. This type of analysis avoids the necessity of prior decisions about the FSC and SSC characteristics of the cells of choice (e.g., lymphocytes). It becomes particularly important when the cells of interest are less homogeneous in physical characteristics than lymphocytes. For example, by gating on a stain that is speci®c for cytokeratin (a protein found on tumor cells of epithelial origin), tumor cells within a mixed population from a breast tumor biopsy specimen can be selected for further analysis (e.g., DNA content) without any prejudgement about the FSC or SSC of a poorly de®ned and heterogeneous population of abnormal cells.
Leukocytes, Surface Proteins, and the Strategy of Gating |
111 |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
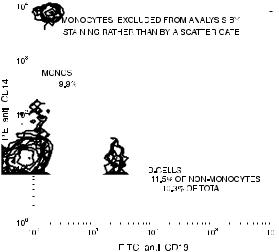
Fig. 6.16. Rather than a scatter gate, a PE-anti-CD14 stain can be used to exclude monocytes from a count of B and non-B lymphocytes. Data courtesy of Jane Calvert.
A logical extension of this kind of technique can be seen in the methodology proposed by PK Horan in 1986: No decisions based on the scatter characteristics of cells have been made. Cells are stained simply with a cocktail of conjugated monoclonal antibodies at appropriate (sometimes not saturating) concentrations, and all the cells in the mixture are classi®ed according to their staining characteristics (Fig. 6.17). Staining cells from leukemic patients currently follows a similar strategyÐwhere abnormal and normal cells are de®ned by the way they cluster in a two-dimensional dot plot. In other words, cells are de®ned by their relative intensities more than by their negativity or positivity for a given antibody.
At the beginning of this section, a strategy was described for placing a scatter (FSC/SSC) gate and then evaluating it in terms of purity and inclusivity. We were then forced to admit that gating is often an uneasy and subjective compromise between these con¯icting criteria. We ®nd therefore that we must conclude that using FSC and SSC characteristics to gate cells may not be a good thing after all. The availability of multicolor analysis has led to a trend toward using staining characteristics to de®ne the cells of interest without regard to

112 |
|
|
|
Flow Cytometry |
||
|
60 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NK cells |
||||
|
|
|||||
|
|
|
||||
intensity |
50 |
|
|
Ts cells |
||
|
|
|||||
40 |
|
|
||||
|
|
|
|
|||
fluorescencered |
30 |
|
Th cells |
|||
|
||||||
|
|
|||||
|
|
|
||||
log |
20 |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
10 |
|
B cells |
|
|
M
10 |
20 |
30 |
40 |
50 |
60 |
log green fluorescence intensity
Fig. 6.17. The two-color ¯uorescence pro®le of peripheral blood mononuclear cells stained simultaneously with six di¨erent monoclonal antibodies to delineate ®ve different populations of cells. From Horan et al. (1986).
their possibly variable physical (light scatter) properties. By using one or more colors in the analysis either to select or to exclude (gate in or gate out) particular groups of cells, we can avoid any prejudgement on the physical characteristics of those cells. This overall strategy makes sense when our system allows for multicolor analysis (with parameters to spare) and when antibodies are available to de®ne the cells of interest. It is, nevertheless, still true that antibodies to de®ne subsets of cells are not perfect: Cells of di¨erent phenotypes react with similar antibodies (sometimes, thankfully, at di¨erent intensities), and several antibodies are often required to fully de®ne the taxonomy of a cell.
As a summary comment on gating, we need simply to remember that in ¯ow cytometry our questions are usually formulated in terms of ``what percentage of a certain population of cells is positive for a certain set of characteristics?'' The choice of a gate de®nes that ``certain population'' of cells. The choice of that gate will therefore a¨ect the answer to the question (the percentage positive). Whether gating
Leukocytes, Surface Proteins, and the Strategy of Gating |
113 |
is applied by means of scatter characteristics, by means of staining characteristics, or not at all, the procedure still needs to be described and quanti®ed if the results are to be meaningful and reproducible. It is only when we have stated exactly which ``certain population of cells'' we are analyzing that we have ful®lled our goals of objectivity and/or explicit subjectivity in ¯ow analysis.
FURTHER READING
Chapters 3 and 5 in Ormerod, Chapters 3.2 and 3.3 in Diamond and DeMaggio, Chapters 10 and 11 in Darzynkiewicz, Chapter 6.2 in Current Protocols in Cytometry, and Chapters 17 and 34 in Melamed et al. are all good discussions of general lymphocyte staining methodology for ¯ow analysis.
Chapter 1.3 in Current Protocols in Cytometry and Chapter 14 in Darzynkiewicz (both by Robert Ho¨man) are excellent discussions of sensitivity and calibration.
Volume 33, number 2 (1998) of Cytometry is a special issue devoted to ``Quantitative Fluorescence Cytometry: An Emerging Consensus.''

Flow Cytometry: First Principles, Second Edition. Alice Longobardi Givan
Copyright 2001 by Wiley-Liss, Inc.
ISBNs 0-471-38224-8 (Paper); 0-471-22394-8 (Electronic)
7
Cells from Within:
Intracellular Proteins
Although many applications of ¯ow cytometry involve the staining of cells for proteins expressed on the outer membrane, cells also have many proteins that are not displayed on their surface. With appropriate procedures, ¯ow cytometry can provide a means to analyze these intracellular proteins. The outer cell membrane is impermeable to large molcules like antibodies; however, if we intentionally ®x cells to stabilize proteins and then disrupt the outer membrane, the cells can be stained with ¯uorochrome-conjugated monoclonal antibodies against intracellular proteins. After time to allow the antibodies to pass through the now-permeabilized membrane, the cells are washed to remove loosely bound antibodies and then are run through the ¯ow cytometer to measure their ¯uorescence intensity.
This intensity should, under good conditions, be related to the amount of the intracellular protein present. However, in describing our ability to stain cells for surface proteins, we mentioned that it is best to stain viable cells. Dead cells have leaky outer membranes; they often show high nonspeci®c staining because antibodies get through the disrupted membrane and become trapped in the intracellular spaces. Therein lies a con¯ict in our ability to stain cells for intracellular proteins. Because antibodies of all types are easily trapped in the cytoplasm, there is greater potential for nonspeci®c staining of permeabilized cells than intact cells. The very procedure that we carry out to give access of the staining antibody to its target (intracellular) antigen actually increases the access of all antibodies to nonspeci®c targets. To lower this nonspeci®c background, antibody titers are critical and washing steps are important. Unfortunately,
115
116 |
Flow Cytometry |
even with low antibody concentrations and careful washing, background ¯uorescence from isotype-control antibodies is often considerably higher on permeabilized than on intact cells.
There is, in addition, a second problem. The procedures used for ®xing and permeabilizing cellsÐto give the staining antibodies access to intracellular proteinsÐcan modify or solubilize some antigens, thus destroying the stainability of the very proteins that are being assayed. To make matters worse, the protocol that works best for one antigen may entirely destroy a di¨erent antigen. This should not be surprising after consideration that ``intracellular'' includes proteins of many types and in many di¨erent environments. Some intracellular proteins are soluble, some are bound to organelle membranes, and some are in the nucleus. Therefore, methods for staining cells for intracellular proteins cannot be as standard or as dependable as the methods for staining surface proteins. They have to be individually optimized for the cells and the proteins in question.
METHODS FOR PERMEABILIZING CELLS
While not attempting to describe possible methods in detail, I feel it is important here to point out the issues involved in intracellular staining because they highlight some general issues that a¨ect all of ¯ow cytometric analysis. Methods for permeabilizing and ®xing cells are various and must be optimized for the particular intracellular antigens being detected because some antigens are more robust than others in the face of di¨erent agents. Figure 7.1 gives an example comparing ®xation/permeabilization e¨ects on two di¨erent intracellular antigens: Five di¨erent ®xation/permeabilization protocols have been used, and their e¨ects on staining PCNA (proliferating cell nuclear antigen) and p105 (a mitosis-associated protein) have been compared. The good news is that you can stain for intracellular antigens. The bad news is that it may be di½cult to stain cells optimally for two di¨erent antigens at the same time (and the relative intensity of staining for two di¨erent antigens may tell you little about the actual relative proportions of these proteins in the cell).
The general protocol for intracellular staining involves, ®rst, staining the cells for any surface (outer membrane) antigens, as described in the previous chapter. Then the surface proteins with their

|
|
Intracellular Proteins |
117 |
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 7.1. The e¨ects of di¨erent ®xation protocols on the relative amounts detected of two di¨erent intracellular proteins. Modi®ed from A McNally and KD Bauer as published in Bauer and Jacobberger (1994).
bound antibodies, as well as the intracellular proteins, are ®xed gently to stabilize them. The purpose of the ®xation is to cross-link the proteins well enough that they are not removed or washed out of the cells after the cells are permeabilized, but not so well that the intracellular antibody binding sites are masked or destroyed. Although ethanol and methanol can be used for ®xation (by themselves or following another ®xative), the most common ®xative used prior to intracellular staining is formaldehyde. Formaldehyde is generally used at lower concentration and/or for a shorter period of time than for routine ®xation of surface-stained cells (where ®xation overnight in 1% formaldehyde is the [optional] last step of the procedure before ¯ow cytometric analysis). Formaldehyde (at 0.5±1.0%) for 10 min is a good suggested concentration and time for cell ®xation, but lower or higher concentrations, for shorter or longer periods of time, might be required.
118 |
Flow Cytometry |
This formaldehyde ®xation does permeabilize the cytoplasmic membrane a bit (formaldehyde-®xed cells are permeable to small molecules), but proteins are often cross-linked too tightly for staining of intracellular proteins with antibodies. Therefore the ®xation step is followed by a permeabilization step. Permeabilizing agents are usually detergents, such as Triton X-100, digitonin, NP40, or saponin, at concentrations of about 0.1%. Combined ®xation/permeabilization reagents are also available as proprietary commercial reagents. With luck, the detergent will open up the cell enough so that the now-®xed proteins are accessible to the antibodies used for staining.
What are the criteria by which we can determine whether a ®xation/ permeabilization procedure has been optimized for an antigen in question? This optimization is, in essence, no di¨erent from optimization of a protocol for surface staining of cells. It is ®rst necessary to maximize the ¯uorescence intensity of cells that are known to possess the intracellular antigen (the positive control); ®xation time and concentration need to be altered in combination with di¨erent detergent concentrations to increase the positive staining. It is then necessary to decrease the background staining (using cells stained with isotype-control antibodies) as much as possible; this is done by trying increasing detergent concentrations and washing the cells thoroughly in bu¨er that contains the detergent. In other words, the goal is to increase the signal-to-noise ratio. Because antibody concentration, ®xative agent, ®xative concentration, ®xation time, choice of permeabilization agent, and concentration of that permeabilizing agent are all variables in this protocol (and the optimal characteristics of each may be di¨erent for di¨erent antigens), staining for intracellular antigens requires some persistence on the part of the investigator. The following examples (in this chapter and in the following chapter on DNA) will demonstrate, however, that it is certainly possible.
EXAMPLES OF INTRACELLULAR STAINING
From the point of view of a ¯ow cytometer, surface, cytoplasmic, and nuclear proteins are similar. The ¯ow cytometer cannot ascertain the location of the source of ¯uorescence. In addition, the nuclear membrane has large enough pores that it provides little or no obstacle to staining once the outer, cytoplasmic membrane has been breached.
Intracellular Proteins |
119 |
Cells have been stained successfully for nuclear proteins related to proliferation (for example, PCNA, Ki-67, and various cyclins, which will be discussed in the chapter on DNA) and to tumor suppression (for example, p53, c-myc, and the retinoblastoma gene product). They have also been stained for proteins bound to interior membrane surfaces (e.g., Bcl-2, multidrug resistance protein [MDR], and P-gly- coprotein), and many strictly cytosolic proteins have been analyzed (like tubulin, hemoglobin, surface proteins that exist intracellularly at various stages of di¨erentiation, and many cytokines).
As an example of one of the more complex biological situations, we can use the staining of cytokines as an illustration. Cytokines are a diverse class of proteins that, in response to cell stimulation, are synthesized and then secreted by leukocytes. For example, when T lymphocytes are stimulated, either nonspeci®cally or by immunological triggers, they begin to synthesize interferon-g in their endoplasmic reticulum, send the proteins to the Golgi apparatus, and then secrete the molecules into the environment for stimulation of neighboring cells. To stain for intracellular interferon-g, the usual technique is to stimulate cells with a biological trigger and then to incubate them with an inhibitor (brefeldin A or monensin) for several hours. These inhibitors block the normal secretion of proteins from the Golgi apparatus and thus allow the cytokine concentration to build up in the cell to levels that are detectable. After the incubation period, the cells are stained for any surface antigens of interest, ®xed brie¯y in formaldehyde, permeabilized with saponin, and, ®nally, stained with a monoclonal antibody against interferon-g.
Figure 7.2 shows an example of the way in which cells can be stained for a phenotypic surface marker (CD8) as well as the intracellular cytokine, interferon-g. The ¯ow data indicate that interferon- g is associated, after PMA-ionomycin stimulation, primarily with CD8-negative cells. More of the CD8-negative than the CD8-positive cells have intracellular interferon-g, and those that have that cytokine have more of it per cell. The tricks in the procedure for staining intracellular cytokines are as much biological as chemical (because the stain is for the end result of a functional process). In addition to a knowledge of how to ®x and permeabilize a cell and how to avoid nonspeci®c staining, we require knowledge of how to trigger the cytokine production, knowledge of the time course of cytokine synthesis after stimulation, and knowledge of how long cells can survive