

Metal-Catalysed Reactions of Hydrocarbons / 10-Hydrogenation of the Aromatic Ring
.pdfHYDROGENATION OF THE AROMATIC RING |
457 |
10.2.7. Hydrogenation of Benzene to Cyclohexene85
As noted in Section 10.12, the partial hydrogenation of benzene to cyclohexene is an economically attractive first step in the synthesis of polyamides, because it is more easily separated from unreacted benzene than is cyclohexane. The reaction is however a difficult one to accomplish, because cyclohexene is normally much the more easily hydrogenated,86 so its appearance in any significant amount depends upon constructing a catalyst which is in effect selectively poisoned for the hydrogenation of the alkene but not for the more strongly adsorbed benzene. There have been many attempts to modify metals (mainly ruthenium87) to secure this result, in ways that are now briefly recounted.
Many of the available results have been obtained in three-phase systems, using autoclaves at 423–473K and 10–70 atm hydrogen;88 unsupported ruthenium catalysts have often been used, sometimes generated in situ by precipitation of the hydroxide from an aqueous solution RuCl3, followed by its reduction on introduction of hydrogen.87,89 All agree that the presence of water is essential in securing reasonable selectivity to cyclohexene, but its role is not well understood.88 Early results indicated that iron salts arising from corrosion of the stainless steel of the autoclave might be beneficial to selectivity, although inimical to rate.89 Many salts have subsequently been tried, the sulfates of iron, cobalt and zinc proving best;90,91 in these cases, selectivities of about 50% have been obtained, at a sacrifice of two-thirds of the rate. The role of the salts may be to render the metal’s surface hydrophilic and thus to secure better access of water to the surface,90 and so fulfil its role, whatever that is. Various bases have been used to precipitate the Ru(OH)3, those of calcium and strontium being best (selectivities respectively 64 and 67%).87 Various supports for ruthenium have also been tried,92 those of ytterbium, zirconium and iron (FeIII), with potassium or calcium hydroxide, all giving selectivities over 70%.87
A practical large-scale process would be better conducted in a continuous mode, for which vapour-phase reaction is preferable, so this has attracted some attention. The presence of water is still essential, but other modifiers such as ethylene glycol (1,2-dihydroxy-ethane) and -caprolactam have proved beneficial.93 A good understanding of the role of these various parameters and modifiers is not yet available.
Alkyl substitution on the benzene ring increases the selectivity for partial reduction.2,88.,93,94 With toluene and ethylbenzene, using ruthenium with zinc oxide, the 2,3- alkylcyclohexene was surprisingly the main product, this being less stable than the 1, 2- isomer; m-xylene however gave chiefly 1,4-dimethyl-1, 2-cyclohexene. The presence of tert-butyl groups is even more effective in easing the intermediate alkene off the surface: thus even with rhodium 1,2-di- tert-butylbenzene gave 40% of the 1,2-dialkyl-2,3-cyclohexene, while 1,3,5-tri- tert-butylbenzene afforded 65% of the 1,3,5-trialkyl-1,2-cyclohexene.95

458 |
CHAPTER 10 |
Cyclohexadiene is very rarely seen as a product of benzene hydrogenation, but inexplicably the 1,3-isomer constituted about 80% of the initial products when the reaction was conducted in isopropanol with a commercial 1% Ru/C catalyst: smaller but still significant amounts were found with 2.5% Ru/TiO2.96 Metallic impurities in the supports (C, Al2O3, SiO2, TiO2) were found to matter. The 1,4-isomer reacted much more quickly than benzene over Ru/SiO2 and Cu/SiO2, but hydrogenation of a mixture of the 1,3-isomer and 14C-labelled benzene on several metals including ruthenium showed that the label appeared more in cyclohexane than in cyclohexene,97,98 perhaps because the diene was largely excluded from the surface by the more strongly adsorbed benzene.
10.3. HYDROGENATION OF ALKYL-SUBSTITUTED BENZENES
10.3.1. Kinetic Parameters
Although the work described in Section 10.2 related predominantly to benzene, many of the studies described used toluene and the xylenes as well, and because the hydrogenation of all of these compounds had many features in common it would have been inappropriate to separate them. However there are some further aspects which merit attention, and which if introduced earlier might have interrupted the flow of the argument; and also there are results to be mentioned concerning more highly substituted molecules. Some recapitulation of what has been said before is however inevitable in order to provide a complete picture.
It is now well-established that rates of hydrogenation decrease with the number of methyl substituents,99 although with the xylenes and more extensively substituted molecules they depend upon their relative positions1,2,86 (Figure 10.9). For the former the sequences of rates or TOFs on Ni/SiO2,4,5 Raney nickel and Adams platinum1,2 is para > meta > ortho. There is general agreement that the cause of the reactivity hierarchy is to be sought in the electron-releasing effect of the substituents, which increases the electron density within the ring in proportion to their number, and thus leads to stronger π -donor bonds to the surface. This effect is demonstrated by a decrease in ionisation potential, and to an increase in stability constants for the formation of charge-transfer complexes,12 as well as in the heats of adsorption deduced from the kinetic analysis of the reactions on Ni/SiO2.4,5 Although it might explain a decrease in the rate of exchange (for which quantitative results are lacking), it is by no means clear that it would affect the ease of attack on the ring, and the variations of rate with isomer structure argue for a geometric as well as an electronic effect. Relative rates for n-alkyl substituents of increasing chain length reach a lower limit after four carbon atoms,1,2 and molecules with secondary and tertiary substituents are less reactive than corresponding methyl compounds.86 Pentaand hexamethyl-benzene are very difficult to hydrogenate.1

HYDROGENATION OF THE AROMATIC RING |
459 |
Figure 10.9. Effect of n- (—O) and sec- (—) alkyl substituents on the rate of hydrogenation of the aromatic nucleus on platinum.2
Apparent and derived true activation energies increase (and those obtained above Tmax become more negative) as the number of substituents increases when Ni/SiO2 is used,4.5 but the effect is not the same with all catalysts.12 The similarities in the temperature dependence of the reaction orders suggests4,5 that the tightness of packing of aromatic molecules on the surface is not much affected by inserting one or two methyl groups, and the energetics of binding to the surface is indeed the source of the reactivity differences.
The difference between benzene and toluene has given a group of French scientists the bright idea of using their relative reactivities, or more properly the relative values of their adsorption coefficients (KT /K B ) derived therefrom, as a way of seeing how the electron-deficient character of surface metal atoms depends upon prevailing circumstances.100 The more electron-deficient the surface, the more strongly should toluene be adsorbed relative to benzene, and thus its reactivity should be diminished relative to benzene: in other words KT /K B should measure the number of unoccupied states in the metal’s d-band.100,101 Thus this ratio has been shown to decrease dramatically on passing from Group 8 to 9 to 10 (Table 10.4), and its values (except for that of ruthenium) run parallel to the electronic specific heat, which in turn depends on the density of states at the Fermi surface.100 Two examples of the application of this concept may be cited: (1) with
TABLE 10.4. |
Ratio of the Adsorption Coefficients for Toluene (KT ) and Benzene (KB ) |
||||||
|
|
|
on the Noble Metals of Groups 8 to 10 |
|
|
||
|
|
|
|
|
|
|
|
|
|
Ru |
Rh |
Pd |
Os |
Ir |
Pt |
|
|
|
|
|
|
|
|
KT /KB |
200 |
10 |
1 |
55 |
24 |
8 |
|

460 CHAPTER 10
Pt1−x Zrx supported on carbon or alumina, KT /K B is proportional to x , suggesting electron transfer from platinum to zirconium, as predicted by the Engel-Brewer theory, and (2) chemisorption of sulfur on platinum has been shown to decrease electron density of the surface, while carbon has the opposite effect.102 The ratio KT /K B was very large for ruthenium, about 10 for rhodium and about unity for palladium,85 which may help to explain their different activities in these and other reactions. An extensive kinetic study of the hydrogenation of mixtures of benzene and toluene on Ni/Y zeolite has however revealed a situation of some complexity,33 and it is not certain that the original simple concept is totally valid.103
10.3.2.Stereochemistry of the Hydrogenation of Alkyl-Substituted Benzenes6,104
It has been known since 19222,105 that the principal product formed by the hydrogenation of dialkylbenzenes is usually the corresponding Z - dialkylcyclohexane, although the amount of the E -isomer depends upon a number of factors. (1) With the xylenes, it varies with the isomer,1,53,88,106,107 and with rhodium, ruthenium and platinum the sequence is para > meta > ortho. (2) Its proportion increases with temperature2 and with hydrogen pressure36,108 in each case. (3) Rhodium affords more of the E -isomer than does ruthenium.2 These observations relate to experiments performed with liquid or solution, and other factors have also been recognised.2 In the gas-phase on Ni/SiO2, the E -selectivities for each isomer vary with temperature as shown in Figure 10.10;53 it may be more than coincidence that o-xylene achieves the same value as the temperatureindependent value shown by m-xylene, but no explanation has been suggested. Ru/Al2O3, Ru/SiO2 and Ru/TiO2 all gave 10–20% of the E -isomer from o-xylene, its amounts decreasing with increasing particle size.109
Figure 10.10. Temperature-dependence of the selectivity to E-dimethylcyclohexane in the hydrogenation of each of the xylene isomers over Ni/SiO2 .53

HYDROGENATION OF THE AROMATIC RING |
461 |
There have been lengthy discussions concerning the significance of these observations, without firm conclusions being reached.53,106 Epimerisation of the cyclohexane is clearly not a viable general explanation,53 and the phenomena are obviously related to those seen with alkylcyclohexenes, discussed in Chapter 7. Stereochemistry is presumably decided as the final pair of hydrogen atoms is added, so that Z -addition, as with the cyclohexenes, is not unexpected; but the considerable formation of the E -isomer may require the temporary desorption and re-adsorption of one of the intermediates,110 as was suggested previously (Section 7.5.3).
10.4.HYDROGENATION OF MULTIPLE AROMATIC RING SYSTEMS
10.4.1. Polyphenyls
This short section is concerned with molecules having two or more benzene rings that are not condensed. The rings therefore have a degree of independence from each other, and so their reduction may show some selectivity.
Biphenyl has been hydrogenated on five of the noble metals supported on carbon, and palladium again distinguished itself by showing 100% selectivity to cyclohexylbenzene: the others gave values between 50 and 65%.94 It also excelled in reducing diphenylmethane to cyclohexylphenylmethane with 89% selectivity.111 Over Adams platinum, m-terphenyl suffered selective reduction of the central ring, but with the para-isomer it is the terminal ring that was reduced first.88
Many years ago, Homer Atkins and his colleagues examined the reduction of triphenylmethane catalysed by nickel powder;112 it went non-selectively to the tri-cyclohexylmethane. 1,3,5-Triphenylbenzene was reduced to the tricyclohexylcyclohexane. With phenylethanes, rates decreased slightly with increasing number of phenyl groups.113
10.4.2. Fused Aromatic Rings: (1) Naphthalene94,114
This section is concerned with the hydrogenation of naphthalene and of tetralin (tetrahydro-naphthalene) and their alkyl derivatives, and the reactions with deuterium. The focus of interest lies in the stereochemistry of the reactions, in the analysis of the products (sometimes very complicated), and their significance for the understanding of reaction mechanisms.
There is much evidence from early work summarised by H.A. Smith,2 and amply confirmed by later careful work,114 that naphthalene can be hydrogenated to tetralin, i.e. one of the rings could be saturated, with a high degree of selectivity on nickel and the noble metals (Table 10.5), although in some cases it declined somewhat at higher temperatures. In further hydrogenation, yields of octalin were

462 CHAPTER 10
TABLE 10.5. Selectivities in the Hydrogenation of Naphthalene to Tetralin (ST ), Octalins (SO ) and Decalin (SD ) on Metals of Groups 8 to 10114
Metal |
Support |
T/K |
ST |
SO |
SD |
Ru |
Al2 O3 |
303 |
82.0 |
1.9 |
16.1 |
Rh |
Al2 O3 |
298 |
95.1 |
0.9 |
4.0 |
Pd |
Al2 O3 |
373 |
99.8 |
— |
0.2 |
Ir |
C |
298 |
86.5 |
0.4 |
13.1 |
Pt |
Al2 O3 |
473 |
96.3 |
0.2 |
3.5 |
low because of the fully saturated decalin. It is natural to enquire how the two initially identical rings differ when they interact with the surface to account for this highly selective partial reduction.115 There are no structural studies to guide us, nor indeed are there any on other compounds having two or more rings (e.g. biphenyl) that might delineate the separation of adsorption sites. It has however been suggested114 that there are two possible forms, in one of which the interaction is through only one of the rings, and in the other of which it is through one (or perhaps two?) of the C C double bonds represented in one of the canonical forms (Figure 10.11). It must also be remembered that the total resonance energy in naphthalene (255 kJ mol−1) is less than twice that of benzene, so that the second ring will be harder to reduce than the first.
C double bonds represented in one of the canonical forms (Figure 10.11). It must also be remembered that the total resonance energy in naphthalene (255 kJ mol−1) is less than twice that of benzene, so that the second ring will be harder to reduce than the first.
The presence of a single alkyl substituent in the 1-position has a marked effect on which ring is reduced first: with Pd/C as catalyst, the proportions of reduction of the ring carrying the substituent increase in the sequence: methyl (34%) < ethyl (45%) < isopropyl (68%) < tert-butyl (97%).88 With platinum, however, the unsubstituted ring is the more reactive.114 The size and position of the substituent also influences its rate of reduction. There are no kinetic studies to differentiate between effects on the adsorption coefficients and on the rates of reaction of the molecules once they have been adsorbed, of the kind conducted with benzene and toluene; a combination of steric and electronic factors may be at work.
Figure 10.11. Representations of the adsorbed states of naphthalene.114
(A)Interaction with the surface through one ring.
(B)Interaction through one (or two) C C double bonds in a frozen canonical form (the likely form on palladium).
C double bonds in a frozen canonical form (the likely form on palladium).
HYDROGENATION OF THE AROMATIC RING |
463 |
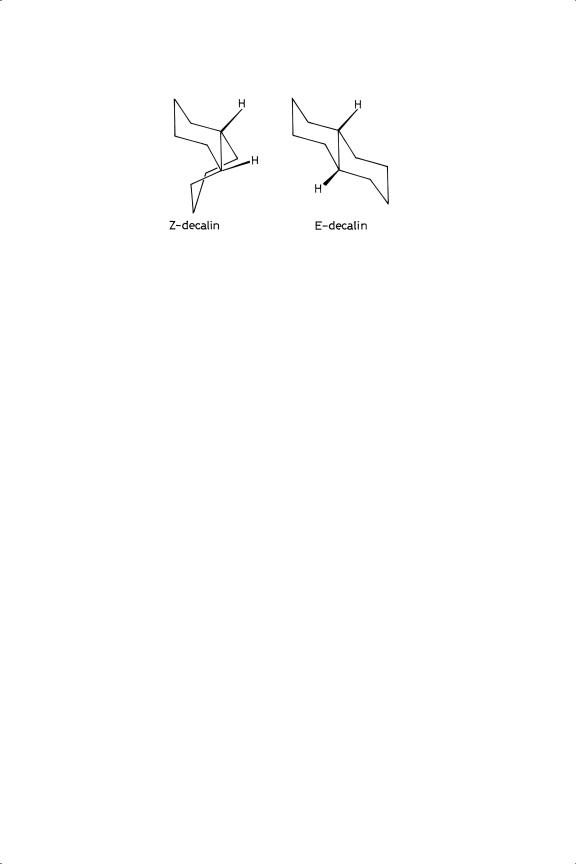
Figure 10.12. Structures of Z - and E-decalin.
Decalin exists as Z - and E -isomers (Figure 10.12), and if all ten hydrogen atoms were added to the same side of the naphthalene ring Z -decalin would be the sole product. In fact both products are formed;114 ruthenium showed very high selectivity to the expected Z -product, while palladium gave about 50% of the E -isomer, the other noble metals giving intermediate values. Palladium was also notable for producing more than 80% of the E -isomer from 1,9-and
1,10-octalins (Section 7.53).114 Selectivities to the two isomers were about the same when tetralin was the reactant. The mechanism whereby the E -isomer is formed has been the subject of much discussion, and the problem is closely related to that of E -addition to substituted cycloalkenes (Section 7.5.3). The possibility that the E -isomer arises from topside reaction of a hydrogen (or deuterium) molecule has been dismissed for a variety of reasons, so the remaining option, namely, that of desorption, inversion, and re-adsorption of an octalin intermediate, has received careful consideration.114 This hypothesis requires it to be shown that octalins are indeed formed, that they are sufficiently reactive, and that they in fact do what is expected of them. Of the six possible octalin isomers, the 1,9-predominates, and it is Z -addition of this isomer that leads to E -decalin114 (Scheme 10.3). Table 10.5 indicates that small amounts of octalins are indeed formed and are reactive in the presence of the aromatic compound; in fact they were initially very significant products (15–30%) of the hydrogenation of both naphthalene and tetralin, but especially with the former their concentration quickly decreased as reaction proceeded. Palladium is noted for its ability to hydrogenate alkenes in the presence of aromatics, while ruthenium, rhodium and iridium favour aromatics over alkenes. This has been nicely demonstrated by hydrogenating o-xylene with a mixture of palladium and (say) ruthenium catalysts: the latter produced cycloalkenes, which were then rapidly hydrogenated by the former, so that lower amounts of the cycloalkenes and more of the E -dimethylcyclohexane were found than with ruthenium alone. The distinctive properties of palladium are again evident, but the different behaviours of the various noble metals still need an explanation. With palladium and platinum on (or in?) various zeolites, their acidity affected the ratio

464 |
CHAPTER 10 |
Scheme 10.3. Formation of Z- and E-decalins by Z-addition of hydrogen to octalin intermediates in the hydrogenation of naphthalene or tetralin.
of Z - and E -decalins formed from naphthalene, high proportions of either being obtainable by appropriate choice of components: metal particle size was not a determining factor.116
The consequential studies of the hydrogenation of the octalins117 have been reviewed in Section 7.5.3. To cut a very long story short, the suggested route to E -decalin has been validated, or at least has been found to be satisfactory; of course if we believe Karl Popper there may be an even better explanation around the corner.
The addition of a methyl substituent onto the naphthalene nucleus differentiates the two rings: hydrogenation of 1- and 2-methylnaphthalene has been examined on all the noble metals of Groups 8 to 10 (except osmium),114 and with Ru/Al2O3 about 80% of reduction occurred at the unsubstituted ring. (Note the numbering of the atoms in the methyltetralins starts on the reduced ring). Hydrogenation of the methyltetralins revealed another novel feature, namely, their isomerisation in parallel with their reduction: thus hydrogenation of 1-methyltetralin at 473K and 55% conversion with Pd/Al2O3 gave 38% 5-methyltetralin, 2% octalins and 60% decalins. The other metals gave small amounts (<4%) of the isomer, and similar but less marked effects were seen with 2-methyltetralin. There are four stereoisomers of each of the methyldecalins (see Scheme 10.4 for their structures and naming). Except with palladium, where the E-anti-isomer predominated, the Z -syn-isomer was the principal one: ruthenium gave 95% of the two Z -isomers, but palladium only 55%, the others giving intermediate values. It was suggested that re-adsorption of the methyloctalin in its least hindered orientation was slow and structure-determining with ruthenium, whereas formation of the half-hydrogenated state controlled product structure with palladium. Of course, as isotopic labelling experiments have indicated, the alkene may re-adsorb in the same orientation: it

HYDROGENATION OF THE AROMATIC RING |
465 |
Scheme 10.4. Designation of the isomers of 1-methyldecalin.
appeared that the way the methyloctalin re-adsorbed was more affected by the methyl group in the 1- than in the 2- position.
The use of dimethylnaphthalenes114 brought additional complications, but no new principles. There are 68 geometric isomers of the dimethyldecalins, but each symmetrically-substituted naphthalene gives only six products, and unsymmetrically-substituted eight: nevertheless their gas-chromatographic analysis required a capillary column 15.2m long, having the equivalent of 105 theoretical plates. Once again, ruthenium and palladium (the only two metals used) had clearly differentiated properties: the former hydrogenated cleanly, with a minimum of desorption and re-adsorption, and because Z -addition predominated it was concluded that the transition state for saturation had a high conformational energy. Palladium on the other hand induces in adsorbed molecules ‘a state of rather violent agitation’, in which hydrogen atoms migrate freely and double-bonds move at will, and desorption-re-adsorption is frequent: on this metal therefore the transition state to the alkane has a somewhat low conformational energy, and so the less stable Z -isomers are major products.
The exchange of naphthalene with deuterium has also been followed on the noble metals of Groups 8 to 10, excepting osmium.118 Stepwise and multiple exchange were detected, the former dominating on palladium, iridium and platinum at 473 K at high deuterium pressure. Each metal however disclosed its individuality. On palladium, for example, the d2- and d4- products were most marked, perhaps because naphthalene, adsorbed in the diene form (Figure 10.11), added two deuterium atoms, then inverted and lost the two hydrogen atoms to revert to naphthalene: reiteration of these steps would give the d4- molecule. Ruthenium and rhodium showed similar behaviour. With platinum, a sharp discontinuity between d4- and d5- molecules suggested that exchange might experience difficulty in proceeding from one ring to the other. The results were interpreted in terms of Burwell’s equilibration model (see Section 8.3.5).

466 |
CHAPTER 10 |
It is necessary to offer a short appreciation on the scope and import of the work reported by A.W. Weitkamp in a very substantial paper,118 a major review114 and a lengthy conference abstract,117 concerning the reactions of hydrogen and of deuterium with naphthalene and its homologues, and tetralin and the octalins and their homologues, inadequately summarised in this section and Section 7.5.3. Perhaps because of its intricacy and complexity, this monumental, detailed and comprehensive study has not received the acclamation it deserves, nor has it been integrated into the corpus of knowledge of the mechanisms of metal-catalysed hydrocarbon reactions. This major project was conducted in the R and D Department of the American Oil Company, and its execution required all the resources of an industrial laboratory: it was beyond the reach of any academic institution, and it is unlikely that such a single integrated set of reactions will ever again be subject to such intensive study. As well as its purely catalytic content, which is vast, the work embraced the identification, conformational analysis and thermodynamic properties of the methyland dimethylhomologues of naphthalene and its hydrogenated products. The skills deployed in achieving the outcome are quite remarkable. Much of what was discovered was not fully explicated, and could usefully be re-examined with the aid of now-available computational procedures. Anyone wishing to develop a new area of research in this sector of catalysis would be well advised to scan these publications, but unfortunately there was little physical characterisation of the catalysts reported, and it seems that the stereochemical variations that these reactions enjoy makes them very suitable for further exploring particle size effects and the behaviour of bimetallics. The very clear differences between ruthenium and palladium are only one of the puzzles that this work gives rise to. Undoubtedly there is here a further major area to explore; someone really should examine naphthalene on single crystal surfaces, especially of palladium. It has to be the task of another generation of scientists to exploit this family of reactions, the better to understand how hydrocarbon transformations occur on metal surfaces.
10.4.3. Fused Aromatic Rings: (2) Multiple Fused Rings1,2,88,111
The hydrogenation of polycyclic aromatic hydrocarbons has long been a subject of great interest, chiefly to organic chemists, but hardly any fundamental studies of mechanism have been reported. Attention has focussed on the ease with which the various rings can be reduced, and on what intermediate partially reduced products can be formed. The structures of some of these compounds are shown in Scheme 10.5; some of them are notoriously carcinogenic.
It is hard to summarise the older literature,1,2 because such a variety of catalysts and conditions was used. With anthracene, the central ring 2 was first reduced, but as further hydrogen atoms were added isomerisation occurred, leading to a product in which only the terminal rings 1 and 3 were reduced.88 Phenanthrene