

Применение неэмпирических и полуэмпирических методов в квантовохимических расчетах - metod421
.pdfНажав последовательно dA, dA, dG, dT мы получим двойную спираль ДНК, выставив предворительно галочку, в опции Double Stranded.
3.2.2 Работа с выделенными атомами (молекулами)
Удалить или копировать сразу несколько атомов или всю молекулу после их выделения кнопкой (select) можно выполнив нескольких команд.
1 Указать галочкой - атом, остатки (residues), молекула в меню Select.
2 После того, как Вы настроили параметры выделения (например, атом), нужно навести курсор на выделяемый объект в рабочем поле и сделать L- щелчок кнопкой мыши.
3 Таким же образом можно выделить связь между двумя атомами. В этом случае в параметрах выделения должен быть обязательно отмечен галочкой
Atoms.
Выделенные фрагменты можно удалить или запомнить в буфер с последующим извлечением из него (соответствующие кнопки на меню или разделы в меню Edit).
Выделенные фрагменты можно также отдельно перемещать или вращать на рабочем поле, выбрав соответствующие инструменты и “схватив” их правой кнопкой мыши (только для этого необходимо в меню
Fail/Preferences/Tool отменить whole molecular translation).
Для отмены выделения поместите курсор в пустой области экрана и щелкните левой кнопкой мыши. Чтобы отменить выделение отдельного атома, фрагмента или связи курсор наводят на выделенный объект и однократно щелкают правой клавишей мыши.
3.2.3 Изображение водородных связей
Чтобы подтвердить благоприятные условия для образования водородных связей, HyperChem вычисляет их и выводит на дисплей.
Водородные связи формируются, если расстояние до водородного донора - менее чем 3.2 Å и угол ковалентной связи донора и акцептора - менее чем 120 градусов.
Чтобы подтвердить условия создания водородной связи необходимо:
1 В меню Display отметьте галочкой пункт Show Hydrogen Bonds (Показать водородные связи)
2 Там же выбрать Recompute H Bonds (Вычислить заново водородные связи)
HyperChem отображает водородные связи пунктирной линией. Водородные связи не вычисляются автоматически в каждой
конфигурации, поэтому, когда Вы изменяете геометрию молекулы, водородные связи приходится изображать и вычислять заново.
3.2.4 Расчет характеристик молекулы в гидратной оболочке
61
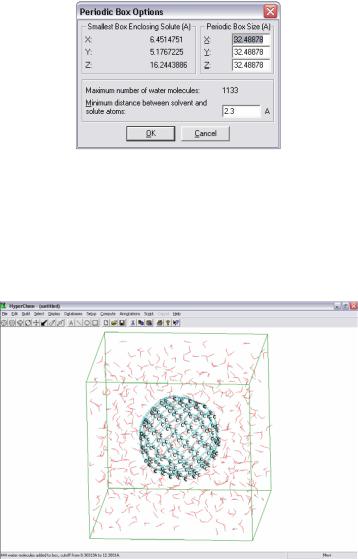
В HyperChem существует возможность поместить молекулу в водную среду. Для этого необходимо воспользоваться командой setup > periodic box.
Рисунок 10 - Задание периодических условий.
После открытия данного диалогового окна задают размеры периодических параметров ящика, который с нажатием кнопки «ок» автоматически заполняется молекулами воды.
Рисунок 11 – Система, окруженная молекулами воды
Указав параметры коробки необходимо отрелаксировать систему, перед тем как сохранить файл, ибо не отрелаксированные химические системы при расчёте некорректно разлетаются. Для этого в setup > molecular необходимо выбрать метод расчета и с помощью опций «Option» и «Components» выставить следующие установки:
62

Рисунок 12 - Параметры для молекулярной динамики
Затем входим в compute > geometry optimization, и выставляем следующие параметры:
Рисунок 13 – Параметры для релаксации
Градиент можно выставить в разумных пределах (например, 0.01). Нажимаем кнопку «Ок». Начиная с этого момента, Сyperchem начнет усиленно считать, пока не достигнет градиента меньше, чем указано, или пока не будет нажата кнопка «cancel».
После окончания расчетов можно сохранить результаты нашего труда. Hyperchem может сохранять в различных форматах, из которых нам понадобятся только два: формат – hin и так называемый protein data bank с расширением ent. Hin обладает тем преимуществом, что в нём сохраняется вся информация, которой располагает НyperСhem, в частности периодические условия, зато ent удобно редактировать текстовым
63
редактором и именно его понимает modyp, точнее программа premd. Обычно сохраняют как в том, так и в другом формате, причём при сохранении в pdb
надо выставить флажки hydrogens и connectivity.
3.2.5 Расчет методами квантовой химии с предварительной оптимизацией методом молекулярной механики
При расчете химической системы, программа HyperChem выбранную часть системы может рассчитывать каким – либо методом квантовой химии, а остальную методом молекулярной механики. Программа позволяет проводить подобные расчеты с использованием всех полуэмпирических и неэмпирических квантово-химических методов. Необходимо помнить, что при оптимизации геометрии системы только выделенная часть атомов будет менять свои координаты в ходе оптимизации. Остальные атомы будут вносить свой вклад лишь как некое статическое поле, генерируемое зарядами на атомах, вычисленными или присвоенными им ранее.
Чтобы производить расчеты в смешанном режиме, необходимо выделить ту часть атомов молекулы, которая будет считаться как квантовомеханическая. В случае, если некоторые из молекул лишь частично выделены, необходимо убедится, что граничные атомы связаны с остальной частью sp3-связями. Убедится в этом, можно используя параметр Extend to sp3 в меню Select, до того, как запускать полуэмпирический расчет. Эта опция распространит выделение рассчитываемой области по всем направлениям до тех пор, пока выделенная часть не достигнет конечного атома молекулы, либо не найдет sp3-sp3 связь.
3.2.6 Расчеты методом Аb initio в рамках пакета HYPERCHEM
Диалоговое окно (рисунок 14) служит для более точной настройки параметров ab initio расчетов.
Опция Integral Format может работать в режиме Regular.
Regular определяет использование обычного формата для записи двухэлектронных интегралов. HyperChem использует 16 байт для записи каждого из интегралов. Первые 8 байт хранят 4 индекса интеграла, а последние 4 – его значение. Программа сохраняет эти величины только в том случае, если абсолютное значение интеграла больше или равно параметру Cutoff. В противном случае значение этого интеграла приравнивается к 0. Двухэлектронные интегралы и их индексы сохраняются на диске без модификации при выборе опции Regular и могут быть записаны в .log-файл при соответствующем выборе параметра Quantum PrintLevel меню StartLog.
Параметр Cutoff позволяет сохранять на диске только те интегралы, абсолютное значение которых равно или превышает задаваемый параметр. По умолчанию он равен 10**-10 Хартри. Этот параметр контролирует осуществление SCF(ССП)-итераций, точность волновых функций и энергии,
64

так как он уменьшает количество рассчитываемых двухэлектронных интегралов.
Рисунок 14 – Диалоговое окно для настройки Ab initio Buffer size (Размер буфера)
Данный параметр определяет размер операционной памяти (в словах двойной точности, 8 байт для одного слова), которая требуется для хранения двухэлектронных интегралов до того, как записать их во временный файл на жестком диске (выбор этого диска может быть сделан в меню
File/Preferences/Path).
Достаточно большой размер буфера способен уменьшить расчетное время, поскольку программа будет обращаться к диску менее часто. Если этот буфер будет достаточно велик, то HyperChem не будет обращаться к диску вовсе. Необходимо отметить, что в случае, когда программа останавливается из-за сбоя компьютера, либо по другой некорректной причине, этот временный файл остается на диске. Чтобы освободить это место, его необходимо удалять вручную.
MO initial guess (начального выбора МО)
Параметр определяет начальный выбор коэффициентов молекулярных орбиталей при помощи диагонализации остовного гамильтониана. При выборе параметра Projected Huckel, эти коэффициенты определяются по методу Хюккеля. Аналогично определяются и первоначальные коэффициенты при выборе параметра Projected CNDO (методом CNDO) и Projected INDO (методом INDO).
Number of d Orbitals (Количество d-орбиталей).
Этот параметр определяет вид d-орбиталей, используемые в расчете. Выбор пяти (five) орбиталей соответствует расчету с использованием эрмитовых орбиталей (d 0, d 1, d –1, d 2, d –2), а выбор шести (six) –
65

соответствует расчету с использованием шести d-орбиталей в декартовом представлении (d xx, d yy, d zz, d xy, d xz, d yz).
3.2.7 Построение кривой сечения поверхности потенциальной энергии (ППЭ) на основе расчетов HyperChem
При проведении расчетов в HyperChem можно построить график зависимости расстояния (угла) от полной энергии и найти точки минимума. Для этого формируем систему, выбираем метод расчета в меню Setup, выставляем мультиплетность и заряд системы. После выбора метода расчета, необходимо обязательно выделить связь (или угол), которую будем изменять.
Затем зайти в compute > potential. В строчке Inititial Bond Length нужно указать начальную длину (или угол), а в строчке Final Bond Length нужно отметить конечную длину (или угол), в графе Step задать шаг. В Outliers – различие между энергиями связи, не должно превышать указанного значения. При нажатии «Ok» указанные условия вступят в силу.
На рисунках 15, 16, 17 представлены диалоговые окна, последовательность которых будет появляться при назначении команд, позволяющих рассчитать электронные характеристики комплекса и построить кривую сечения потенциальной поверхности энергии (ППЭ).
Рисунок 15 – Выбор метода расчета
Рисунок 16 – Выбор параметров для построения графика
66

Через некоторое время, необходимое для расчета, на экране появляется график сечения кривой ППЭ (рисунок 17). Если внизу под графиком клекнуть правой кнопкой мыши, то возникнет опция Properties, используя которую можно изменить параметры осей координат и увидеть свойства рассчитанной кривой.
Рисунок 17 – Оформление и свойства графика
Команда System позволяет сохранить данный график. Используя другие опции диалогова окна можно редактировать данный график.
3.2.8 Расчет колебательного спектра молекул
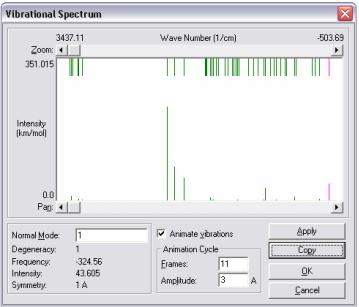
Для того, чтобы рассчитать колебательный спектр молекулы, необходимо предварительно получить расчет, используя любой из квантово-химических методов HyperChem. Затем, в главном меню Compution выбрать опцию vibrations. По окончанию расчетов, вновь зайти в меню «Compution» и осветить Vibrational Spectrum. На экране возникнет ряд полос соответствующих колебательным модам. Номер и частота колебательной моды в см-1 выводятся автоматически в нижней части панели. Здесь же можно выделить интересующую моду и просмотреть ее интенсивность в численном виде, а не только визуально как высоту линии (рис. 18). Поставив галочку в Animate vibrations и нажав «ok» получим на экране наглядное изображение колебаний атомов, соответствующих предварительно отмеченной моде. Вверху шкалы отмечены всевозможные колебательные переходы. Внизу шкалы изображены только разрешенные переходы.
Нажав Copy в диалоговом окне, можно перенести данную картинку на предварительно открытую страницу Word. С помощью вертушек, расположенных в верхней и нижней левой части окна, можно растянуть спектр или сместить его в другую область частот.
Необходимо помнить, что если вы закрыли HyperChem или перешли к другому расчету, то наглядное изображение спектров или МО вернуть невозможно. Картинку можно получить сразу после расчета. Все результаты,
67
полученные машиной, можно сохранить в численном виде, создав перед расчетом log файл. Для этого необходимо выполнить в меню File > Start Log.
Рисунок 18 – Колебательно – вращательный спектр
68
4 Ab initio расчеты в рамках пакета GAMESS
Термин ab initio означает неэмпирическое (непараметрическое) рассмотрение молекулярной системы при решении уравнения Шредингера и полученных на его основе уравнений Рутаана (см. раздел 1.1). В действительности дело обстоит не совсем так. Методы ab initio содержат ряд допущений, облегчающих решение многоэлектронных уравнений. Неэмпирические расчеты более полные и позволяют достичь приемлемой точности, однако на практике ограничения, связанные с объемом памяти ЭВМ и ресурсами машинного времени, позволяют рассчитывать лишь небольшие молекулярные системы.
Во многих случаях ограниченные ресурсы приводят к необходимости уменьшения размеров модельных расчетных систем, при этом важным условием является сохранение определяющих фрагментов. Упрощение решения задачи, например, уменьшение базиса или отказ от учета электронной корреляции могут привести к значительным ошибкам.
Все неэмпирические методы на первой стадии включают однодетерминантный расчет по методу МО ЛКАО ССП (см. раздел 1.1).
Неэмпирические методы расчета, так же, как и полуэмпирические, используют приближение Борна—Оппенгеймера, согласно которому ядра системы остаются неподвижными во времени, а перераспределение электронной плотности для каждого фиксированного положения ядер происходит мгновенно. Это равносильно допущению об отсутствии взаимодействие между электронной и колебательной волновыми функциями, то есть независимости волновой функции электронов от движения ядер. Данное приближение выполняется во многих случаях и вносит лишь незначительные погрешности в рассчитываемые характеристики молекулы. Исключения составляют системы, у которых потенциальная поверхность имеет очень пологий характер, например ян-теллеровские системы.
4.1 Краткая история GAMESS
GAMESS был собран из нескольких существующих квантовохимических программ. Основу составила программа HONDO 5. Для вычисления градиентов энергий ВФ с большим угловым моментом - HONDO 8. Для базисных sp функций - GAUSSIAN76 и GAUSSIAN80. В дальнейшем возможности программы были расширены. Кроме того, копии источника программы стали доступны широкому кругу пользователей. Существующая версия программы подверглась многим изменениям, что продолжается и теперь в Государственном университете в штате Айова США.
В конце 1987 корпорации NDSU и IBM достигли соглашения о развитии исследовательских программ. Результатом совместных разработок явилась версия GAMESS для IBM 3090.
69
Второй этап закончился в 1990 г., когда были существенно увеличены научные возможности GAMESS. Эти дополнения включали вычисления аналитическим методом (ECPs) Гессиана, MP2, спин-орбитального взаимодействия (СОВ), расчет излучательных переходов и так далее. В 1990 году были расширены графические возможности программы и созданы модификации под Х – Windows. Эти программы, теперь управляемы под оболочками Digital Unix или VMS windowing и многими другими. В 1991 году была осуществлена возможность программы визуализировать молекулярные структуры, орбитали, и электростатический потенциал. С 1 июля 1992г., развитие GAMESS продолжилось в Государственном университете штата Айова в лаборатории Ames. В 1996 году было расширено использование существующих параллельных методов и развиты новые. В 1997-1998 гг. Бретт и Бод добавили новые возможности, позволившие аналитически вычислять вторые производные. Были внесены изменения в манере входа в базисные наборы и атомные координаты, включая форму Z- матрицы. Прямое ССП (SCF) было разработано в NDSU. Процедура сходимости была осуществлена Брендой Лам (в университете Хьюстона), для RHF и функций UHF. Модуль CI базируется на унитарных групповых программах Brooks и Schaefer , а также модифицированном методе Davidson используемом GAMESS. Написанный Эльбертом Метод валентных связей (GVB) представляет собой сильно измененную версию GVBONE. Программирование аналитического вычисления CI градиента было сделано Саймоном Вебб в Государственном университете Штата Айова. FULLNR и FOCAS MCSCF программы были пожертвованы фирмой IBM из HONDO программы. Вычисление второго порядка SCF было осуществлено Чабэн (Galina Chaban) в Государственном университете Штата Айова. В 1996 г., Саймон Вебб добавил возможность расчетов с замороженным остовом. Метод MP2 был приспособлен от HONDO. В январе 1999 года, в Государственном университете на Севере Дакоты, Йенсен (Jensen) осуществил возможность вычисления полуэмпирическими методами PM3, AM1, и MNDO, вошедшими в пакет программ MOPAC, как часть GAMESS.
4.2 Возможности программы GAMESS
Программа GAMESS версии 6.0 обладает широким диапазоном возможностей для проведения квантово - химических вычислений.
1.Вычисляет RHF, UHF, ROHF, GVB, или MCSCF.
2.Вычисляет CI или MP2 на основе ВФ полученных при расчете SCF.
3.Вычисляет полуэмпирическими методами MNDO, AM1 или PM3 в
рамках RHF, UHF, или ROHF.
4.Вычисляет аналитические градиенты энергии для всех ВФ, полученных при расчете SCF, плюс MP2 или CI для закрытой оболочки.
5.Оптимизирует молекулярную систему, используя вычисленную энергию градиента, в декартовых или внутренних координатах.
6.Исследует седловые точки потенциальной поверхности энергии.
70