

21
Хартри–Фоку, так как в основном состоянии эти АО не заселены электронами. Поэтому параметры поляризационных функций определяют при расчетах молекул.
Z–терминология применяется и для поляризационных функций. Так, DZP означает двухэкспоненциальный базис, в который дополнительно включены поляризационные функции, TZP – трехэкспоненциальный базис плюс поляризационные функции и т.д. Иногда указывают число наборов поляризационных функций: ТZDP (или TZ2P) означает трехэкспоненциальный Z–базис плюс два набора поляризационных функций. Для молекул, включающих атомы от Li до F, в качестве поляризационных функций обычно используют шесть d–ОГТ, для атома водорода – три р–ОГТ.
Диффузные функции важны для правильного описания анионов и слабых связей (например, водородных связей), для вычислений дипольного момента, поляризуемости, расчетов систем в возбужденных электронных состояниях и т.д. Это обычно гауссианы s– и p–типа с очень маленькими экспонециальными множителями, медленно спадающие с увеличением расстояния от ядра.
12.6. Базисные наборы Попла
Иное соглашение было принято Дж. Поплом с сотрудниками, внесшими огромный вклад в развитие неэмпирических расчетов. Обозначения n–ijG или n–ijkG расшифровываются так: n – число ОГТ для внутренних оболочек; ij или ijk – число ОГТ для СОГТ в валентных оболочках; ij обозначения описывают валентные DZ наборы, ijk – валент-
ные TZ наборы. TZ и DZ называются также валентно–расщепленными базисными наборами.
В базисных наборах Попла число гауссиан s– и p–типа для той же самой sp–электронной подоболочки одинаково, и они имеют одинаковые экспоненты. Однако коэффициенты разложения для СОГТ s– и p–типа различны. Базисные наборы Попла могут также быть расширены за счет включения поляризационных функций d–типа для неводородных атомов (n–ijG* или n–ijkG*) и p–функций для атомов водорода (n–ijG** или n–ijkG**). Так, в молекуле метана СН4 базис 4–31G** предполагает наличие 4–х ОГТ для единственной остовной 1s–АО углерода, 3–х и 1–й ОГТ для каждой из двух функций, аппроксимирующей валентные (2s, 2px, 2py, 2pz) АО углерода (DZ базис), плюс 6 поляризационных 3d–АО на атоме С. Каждый атом Н описывается двумя s–функциями и тремя поляризационными функциями р–типа. Полное число базисных функций равно:
{1 (1s) + 2*[1 (2s) + 3 (2p)] + 6 (3d)} + 4*[2 (1s) + 3 (2p)] = 35.
Табл. 7. Некоторые базисные наборы Попла для молекул, содержащих атомы от Н до F
Базисный |
Описание |
Число базисных функций |
||
|
|
|
||
набор |
|
|
|
|
|
|
|
|
|
|
|
Неводород- |
Водород |
|
|
|
ные |
атомы |
|
STO–3G |
Минимальный базисный набор (полуколичественные |
|
5 |
1 |
результаты в больших системах) |
|
|||
|
|
|
|
|
3–21G |
Двухэкспоненциальный базисный набор (более точ- |
|
9 |
2 |
ное представление для валентных орбиталей) |
|
|||
|
|
|
|
|
6–31G* |
Для неводородных атомов добавлены 6 поляризаци- |
|
15 |
2 |
|
онных d–ОГТ (расчеты систем средней сложности) |
|
|
|
6–31G** |
Для атомов водорода добавлены также 3 поляриза- |
|
15 |
5 |
|
ционные р–ОГТ (вычисление энергий связи) |
|
|
|
6–31+G* |
Для неводородных атомов добавлены диффузные |
|
19 |
2 |
ОГТ (системы с неподеленными парами, молекуляр- |
|
|||
|
ные анионы, возбужденные состояния) |
|
|
|
6–31+G** |
Для атомов водорода добавлены 3 поляризационные |
|
19 |
5 |
|
р–ОГТ |
|
|
|
22
При введении диффузных функций используются следующие обозначения: n–ij+G, или n–ijk+G. Это означает, что к стандартному базисному набору для неводородных атомов добавлен диффузный гауссиан s–типа и 3–и гауссиана p– типа: все они имеют одинаковые экспоненты. Наборы n–ij++G, или n–ijk++G получены из предыдущих добавлением диффузного гауссиана s–типа для атома водорода.
12.7. Роль базисных функций при описании свойств молекул
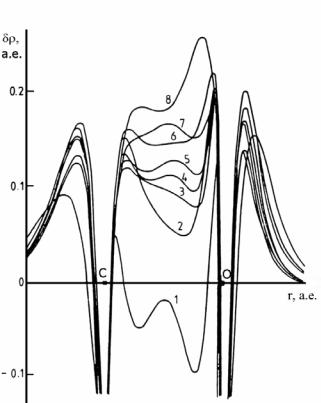
Рисунок 12 демонстрирует, насколько важен правильный выбор базисных функций для предсказания свойств молекул. Малые базисы не в состоянии правильно передать распределение электронов в молекуле СО. причем минимальный базис даже не воспроизводит верный знак дипольного момента. Целесообразность выбора того или иного базисного набора определяется конкретной квантово–химической задачей. Следует иметь в виду, что время расчета главным образом зависит от количества двухэлектронных интегралов, а их число пропорционально числу базисных функций в четвертой степени.
Рис.12. Деформационные электронные плотности δρ(r) молекулы СО (разности между электронной плотностью молекулы и плотностями составляющих ее атомов), вычисленные с использованием следующих наборов базисных функций: 1. SZ;
2. DZ;
3. TZ; 4.DZ+DP; 5. TZ+DP; 6.TZ+2DP;
7 TZ+DP+FP;
8. TZ+2DP+2FP
При определении геометрических параметров молекул хорошие результаты можно получить и в минимальном базисе, при этом ошибки в расчетах длин связей обычно не превышают 0.03 Å и углов – 40. Однако двухэкспоненциальный базис все же предпочтительнее. Расчет силовых постоянных и спектральных характеристик молекул также требует, по крайней мере, использования двухэкспоненциальных базисов. При расчете вращательных и инверсионных барьеров необходимо дополнять базисные наборы поляризационными функциями. Расчет энергий реакций с поляризационными функциями существенно улучшает результат. Для описания водородных связей и межмолекулярных взаимодействий используют двух– и трехэкспоненциальные базисные наборы, расширенные за счет включения поляризационных и диффузных функций для тяжелых атомов и для атомов водорода.

23
При расчете инверсионного барьера учет поляризационных функций играет важную роль. На рисунке 13 представлены потенциальные кривые для инверсионного барьера молекулы аммиака. Видно, что без учета поляризационных функций значение барьера инверсии существенно занижено.
Рис.13. Потенциальные кривые инверсии молекулы аммиака. Расчет проводился ограниченным методом ХартриФока: 1 - в базисе 6-31G*, 2 - в базисе 6-31G
Табл. 8, представленная ниже, может служить некоторым ориентиром, позволяющим выбрать нужный базисный набор на первом этапе квантово–химических вычислений. Данные рекомендации основаны на опыте практических расчетов и в среднем отражают соответствие между рассчитываемыми свойствами и минимальными требованиями к базисному набору.
Табл. 8. Наименьшие базисные наборы, обеспечивающие описание свойств молекул
Свойства |
Базис |
Комментарии |
|
|
|
|
|
Молекулярная геомет- |
|
Исключение – расчет двугранных углов и геометрии пи- |
|
HF/6–31G |
рамидальных структур, где необходимо использовать по- |
||
рия |
|||
|
ляризационные функции |
||
|
|
||
|
|
|
|
Силовые |
HF/6–31G |
Учет поляризационных функций слабо влияет на резуль- |
|
постоянные |
тат в жестких молекулах |
||
|
|||
|
|
|
|
Вращательные и ин- |
|
Исключение – молекулы с осью вращения, пронизываю- |
|
HF/6–31G** |
щей два гетероатома (например, C–N): в этом случае тре- |
||
версионные барьеры |
|||
|
буется базис DZ + Р |
||
|
|
||
Химическая связь. |
HF/6–31G** |
Необходим учет электронной корреляции |
|
|
|
||
Энергии реакций |
МР2/6–31G** |
|
|
|
|
||
|
|
|
|
Взаимодействие ионов |
|
Для расчетов молекулярных анионов и их взаимодействий |
|
и диполей. Водород- |
HF/6–31++G** |
необходимо дополнительно включать диффузные функ- |
|
ные связи |
|
ции |
|
|
|
|
|
Внутри– и межмолеку- |
|
Необходимы как поляризационные, так и диффузные |
|
лярные взаимодейст- |
МР2/6–311+G** |
функции, а также учет энергии корреляции электронов |
|
вия |
|
|
|
|
|
|

24
13. Методы теории функционала плотности
Многоэлектронная волновая функция Ψ очень просто связана с электронной плотностью ρ(r) . Если положе-
ние i–го электрона можно описать оператором локальной плотности eδ(r −ri ) , то электронная плотность есть
N |
|
ρ(r) = Ψ | e∑δ (r − ri |
) |Ψ = |
i |
(50) |
= Ne∫...∫...Ψ0 (r1 , s1 ,..., rN , sN ; R0 ) ×Ψ0 (r1 , s1 ,..., rN , sN ; R0 )dr1ds1..
r1 s1
R0 вектор равновесной ядерной конфигурации.
Электронная энергия системы зависит от электронной плотности основного состояния ρ(r)
E(ρ) = |
∫ |
Vяд(r)ρ(r)dV + |
1 |
∫∫ |
ρ(r)ρ(r ') |
drdr′+G(ρ) , |
||||
2 |
|
|
r −r ' |
|
|
|||||
|
|
|||||||||
|
|
|
|
|
|
|||||
где Vяд(r) – потенциал ядер.
следующим образом:
(51)
Теорема Хоэнберга–Кона утверждает, что существует одинаковый для всех многоэлектронных систем (универ-
сальный) функционал электронной плотности G(ρ). Он представляет собой сумму кинетической энергии и неклассической энергии электрон–электронного взаимодействия, включая обмен и корреляцию электронов. Причем, точная электронная плотность основного состояния обеспечивает минимум функционала (51). Теория, которая изучает способы расчета электронной структуры молекул и кристаллов, основываясь на минимизации функционала (51), называ-
ется теорией функционала (электронной) плотности.
Предположим, что электронная плотность основного состояния взаимодействующих электронов такая же, как и невзаимодействующих. Выражение для кинетической энергии электронов одинаково для всех систем. Тогда мини-
мизация (51) относительно одноэлектронных функций ϕi(r) при условии их ортонормировки и постоянства числа электронов в системе дает уравнения Кона–Шэма:
|
1 |
|
2 |
|
|
|
′ |
|
|
|
|
− |
|
+υˆ(r) + ∫ |
|
|
ρ(r ) |
|
|
dr′+υˆxc (r) ϕi (r) =εiϕi (r) , i = 1,…, m |
(52) |
||
2 |
|
|
|
r −r′ |
|
|
|||||
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|||
где ρ = ∑ϕi 2, υˆxc (r) |
= δExc(ρ)/δρ – обменно–корреляционный потенциал, Exc(ρ) – зависящая от электронной плотно- |
||||||||||
сти обменно–корреляционная энергия. Система уравнений (52) заменяет в теории функционала плотности стандартные уравнения Хартри–Фока. Они решаются самосогласованно, причем все приближения связаны с аппроксимацией обменно–корреляционного потенциала υxc. Все сказанное ранее в отношении использования и выбора базисных функций в неэмпирических квантово–химических методах справедливо и в теории функционала плотности.
Энергии одноэлектронных состояний в теории функционала плотности равны |
|
εi(ТФП)= δE(ρ)/δni , |
(53) |
где ni –электронная заселенность состояния i. В методе Хартри–Фока они равны разности энергий двух состояний с электронными заселенностями, отличающимися на единицу:
εi(ХФ) = EХФ[ni = 1] – EХФ[ni = 0]. |
(54) |
Это означает, что одноэлектронные функции ϕi(r), вообще говоря, отличны от молекулярных орбиталей ϕi(r). Это иногда дает преимущество теории функционала плотности. Например, разложив (54) в ряд Тейлора и приняв во внимание соотношение (53), можно получить для потенциала ионизации весьма точное выражение
Ii ≈ – εi(ni = 1/2). |
(55) |

25
Можно сказать, что потенциал ионизации может быть вычислен с помощью некоего переходного состояния с наполовину уменьшенной электронной заселенностью ВЗМО. Аналогичным образом концепция переходного состояния используется и при расчетах спектров.
Простым, но эффективным вариантом теории функционала плотности является метод Хартри–Фока– Слейтера. Корреляцией электронов здесь, как и в методе Хартри–Фока, пренебрегают, а обменный потенциал представляется в виде
vx(r) = –3α[(3/8π)ρ(r )]1/3. |
(56) |
Величина α лежит в пределах 0,7–1,0 (этот метод часто называют также Xα–методом). Несмотря на кажущуюся про-
стоту, Xα–метод успешно применяется для изучения магнитных свойств достаточно сложных многоатомных веществ, химической связи и др.
Точность, обеспечиваемую Xα–методом, можно оценить, анализируя рис, 14, где представлены результаты расчета различными методами энергий валентных уровней молекулы SF6 в сравнении с данными фотоэлектронной спектроскопии. С экспериментом согласуются лишь неэмпирические и Xα результаты, причем последние значительно точнее (метод CNDO дает неверный порядок уровней), рис. 14.
Рис. 14. Сравнение результатов расчета энергий ионизации валентных состояний молекулы SF6 : а – метод ССП–Хα– РВ (в приближении промежуточных состояний) б – эксперимент ( фотоэлектронные спектры), в – расчет по методу CNDO, г – неэмпирический ССП МО ЛКАО расчет.
14. Полуэмпирическая квантовая химия
Принципиально иное направление, сыгравшее огромную роль в современном развитии химии, состоит в полном или частичном отказе от вычисления одноэлектронных (17) и двухэлектронных (18) – (19) интегралов, фигурирующих в методе ХФ. Вместо точного оператора Фока используется приближенный, элементы которого получают из эмпирических данных. Соответствующие параметры подбирают для каждого атома (иногда с учетом конкретного окружения) и для пар атомов: они либо являются фиксированными, либо зависят от расстояния между атомами. При этом часто (но не обязательно – см. ниже) предполагается, что многоэлектронная волновая функция является одноде-
терминантной, базис минимальным, а базисные функции χiОРТ – симметричными ортогональными комбинациями ОСТ χj . Такие комбинации легко получить из исходных ОСТ χj с помощью преобразования
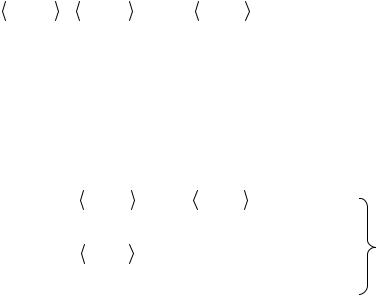
χiОРТ = ∑Sij−1/ 2 χj , |
26 |
(57) |
j
где Sij – элементы матрицы интегралов перекрывания (эта процедура называется ортогонализацией функций по Лев-
дину). Расчет МО проводится обычным итерационным путем.
Полуэмпирические методы работают на несколько порядков быстрее, чем неэмпирические. Они применимы к большим (часто – к очень большим, например, биологическим) системам и для некоторых классов соединений дают более точные результаты. Однако следует понимать, что это достигается за счет специально подобранных параметров, справедливых лишь в пределах узкого класса соединений. При переносе на другие соединения те же методы могут дать абсолютно неверные результаты. Кроме того, параметры часто подбираются таким образом, чтобы воспроизводить те или иные молекулярные свойства, поэтому придавать физический смысл отдельным параметрам не следует.
Основные приближения, используемые в полуэмпирических методах, следующие.
1)Рассматриваются только валентные электроны: считают, что электроны атомных остовов лишь экранируют ядра, поэтому эти электроны учитывают в функциях, описывающих энергию остов–остовного отталкивания (в которое включается ядер–ядерное отталкивание). Поляризацией остовов пренебрегают.
2)В МО учитывают только АО с главным квантовым числом, соответствующим высшим заселенным электронами орбиталям изолированных атомов (минимальный базис), причем считают, что базисные функции образуют набор ортонормированных АО.
3)Для двухэлектронных кулоновских и обменных интегралов вводят приближение нулевого дифференциально-
го перекрывания (НДП):
χ μ* ( r ) χν ( r ) d V = 0 , |
μ ≠ ν . |
(58) |
Считают, что из–за экспоненциальной зависимости радиальной части АО от расстояния еxp(-ζ r) можно
пренебречь двухэлектронными кулоновскими и обменными интегралами, содержащими произведения разных атомных орбиталей:
μν |
|
λσ = μν |
|
λσ δμνδλσ = μμ |
|
λλ . |
(59) |
|
|
|
Это приближение резко уменьшает число вычисляемых двухэлектронных интегралов, поэтому оно в том или ином виде используется во всех полуэмпирических методах.
В приближении НДП, принимаемом для всех пар АО, уравнения Рутана имеют вид:
|
∑ciν (Fμν −εiδμν ) = 0 , |
|
|
μ= 1, 2, 3,…m |
(60) |
||||||
|
|
ν |
|
|
|
|
|
||||
Элементы матрицы Фока записываются следующим образом: |
|
||||||||||
Fμμ = hμμ − |
1 |
Pμμ μμ |
|
|
|
μμ + ∑Pλλ |
μμ |
|
λλ , |
|
|
|
|
|
|||||||||
|
2 |
|
|
|
|
|
λ≠μ |
|
|
|
(61) |
|
1 |
|
|
|
|
|
|
|
|
|
|
Fμν = hμν − |
Pμν μμ |
|
νν , |
μ ≠ν , |
|
||||||
|
|
||||||||||
|
2 |
|
|
|
|
|
|
|
|
|
|
4) Результат расчета не должен зависеть от выбора декартовой системы координат, в которой определяются ориентации p–, d–АО, рис. 15. Приближение НДП нарушает это требование. Такое нарушение вращательной инвариантности имеет место каждый раз, когда двухэлектронные интегралы включают перекрывание двух разных р– и d–АО одного и того же атома. Поэтому в таких случаях в дополнение к НДП часто вводится еще одно приближение: счита-
27
ют, что двухэлектронные интегралы 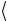 μμ λλ
μμ λλ зависят только от природы атомов, на которых центрированы орбита-
зависят только от природы атомов, на которых центрированы орбита-
ли χμ и χν , и не зависят от конкретного вида орбиталей. Это соответствует сферическому усреднению распределе-
ния валентных электронов на АО различных атомов молекулы при расчете взаимодействия и обеспечивает инвари-
антность решения относительно поворота систем координат. Для усредненного интеграла  μμ λλ
μμ λλ используется
используется
обозначение γAB , где А и В обозначают атомы, на которых центрированы интегралы μ и λ; вычисляется этот усредненный интеграл с s–АО соответствующих атомов
гAB = sA2 |
sB2 |
(62) |
Рис. 15. Иллюстрация происхождения неинвариантности двухцентрового кулоновского интеграла по отношению к повороту осей координат
Что касается одноэлектронных интегралов, то для них также вводятся различные приближения, которые мы рассмотрим ниже применительно к конкретным методам.
Рассмотрим теперь стандартные методы, получившие наибольшее распространение (рис. 16.).
Рис. 16. Основные методы полуэмпирической квантовой химии

28
Табл. 9. Сравнительная характеристика полуэмпирических методов
Метод |
Параметризуемое свой- |
Хорошо воспроизводимые свой- |
Плохо воспроизводимые |
|
(приближение) |
ство |
ства |
свойства |
|
|
|
|
||
|
|
|
|
|
|
Разности энергий |
Дипольные моменты, длины свя- |
Теплоты образования, по- |
|
CNDO/2 |
|
зей, валентные углы, силовые |
тенциал ионизации, сродст- |
|
между занятыми МО |
во к электрону, спектры, ре- |
|||
|
константы |
|||
|
акции |
|||
|
|
|
||
|
|
|
|
|
CNDO/S |
|
|
Теплоты образования, гео- |
|
INDO/S, |
Электронный спектр |
Спектр |
||
метрия молекул, реакции |
||||
|
|
|||
ZINDO |
|
|
|
|
|
|
|
|
|
|
|
Спиновые плотности, константы |
Теплоты образования, по- |
|
INDO |
Спиновые плотности |
тенциалы ионизации, срод- |
||
сверхтонкого взаимодействия, |
ство к электрону, электрон- |
|||
|
|
геометрия молекул |
||
|
|
ные спектры |
||
|
|
|
||
|
|
|
|
|
|
Потенциал атом– |
Теплоты образования, потенциа- |
Электронные спектры, во- |
|
MINDO/3 |
атомного взаимодейст- |
|||
|
вия |
лы ионизации, длины связей |
дородная связь |
|
|
|
|
||
|
|
|
|
|
MNDO |
Теплоты образования |
Теплоты образования, |
Электронные спектры, во- |
|
|
||||
геометрия молекул |
дородная связь |
|||
|
|
|||
|
|
|
||
|
|
|
|
|
АМ1 |
Теплоты образования |
Теплоты образования, |
Электронные спектры |
|
геометрия молекул |
||||
|
|
|
||
|
|
|
|
|
|
Теплоты образования, |
Теплоты образования, геометрия |
|
|
РМ3 |
параметры межмолеку- |
молекул, водородная связь, меж- |
Электронные спектры |
|
|
лярного взаимодействия |
молекулярные взаимодействия |
|
|
|
|
|
|
Рис. 17.

29
На рисунке 17 приведена карта распределения спиновой плотности в молекуле О2, рассчитанная методом INDO. Она показывает, где в этой молекуле локализованы неспаренные электроны. Современные варианты этого метода, дополнительно к обмену, эффективно учитывают корреляцию электронов. Это позволяет рассчитывать с их помощью геометрию и энергии систем с несколькими низколежащими электронными состояниями (ZINDO1) и характеристики спектров УФ и видимого диапазона (ZINDO/S).
Метод MINDO/3 имеет следующие основные недостатки. Теплоты образования ароматических и сопряжен-
ных углеводородов получаются завышенными, а соединений с тройной связью – С≡С– и –С≡N и соединений с неподеленными электронными парами – заниженными. Например, разность рассчитанных и экспериментальных теплот образования молекулы малеинового ангидрида С4H2O3, составляет 114.1 кДж/моль, а для молекул NH2–NH2 и CH3NH–NH2 –149.2 и –105.3 кДж /моль, соответственно. Далее, длины ординарных связей С–С в ненасыщенных углеводородах, С–О в спиртах и С–N в аминах, а также валентные углы С–С=С в алициклических углеводородах, Н–С– Н в метиленах и Н–С–N в аминах получаются с большими ошибками. Геометрия некоторых неорганических молекул (H2О2, H2N2), а также энергия стабилизации димеров предсказываются неудовлетворительно. И, наконец, рассчитанные методом MINDO/3 потенциалы ионизации недостаточно хорошо согласуются с данными фотоэлектронных спектров.
Недостатки метода MINDO/3 преодолены в методе модифицированного пренебрежения дифференциальным двухатомным перекрыванием (MNDO). Схема MNDO следующая. Остовные интегралы hμμ и hμν вычисляются так же,
как в методе CNDO. Параметры βАВ0 для двухцентрового случая зависят только от типа атомов А и В. Атомные пара-
метры βА0 , βВ0 калиброваны по теплотам образования молекул в основном электронном состоянии. Таким образом,
теплоты образования являются параметризуемыми свойствами метода MNDO. Двухцентровые одноэлектронные интегралы VAB вычисляются по формуле
VAB = QA μν |
sB sB , |
(63) |
т.е. АО на атоме А учитываются явно. Двухцентровые двухэлектронные кулоновские интегралы рассчитываются с помощью классических точечных мультиполей на атомах (при этом угловая зависимость всех АО учитывается явно). Чтобы приблизить результат расчета к эксперименту, энергию остов–остовного отталкивания эмпирически завышают. Её определяют по формуле:
EcoreAB = ZAостZBост[ exp(−αA RAB ) +exp(−αB RAB )] sAsA |
|
sB sB , |
(64) |
|
где αА, αВ – параметры метода, ZAост – заряд остова А, R AB – расстояние между центрами атомов А и В.
Метод MNDO хорошо предсказывает теплоты образования и геометрии молекул с ковалентными связями в основном электронном состоянии, но плохо описывает водородные связи, свойства ионных соединений и молекул в электронно–возбужденных состояниях, химические реакции, энергии межмолекулярного взаимодействия. Поэтому были предложены и другие основанные на MNDO, но более эффективные методы: AM1 (Austin Model № 1) и PM3 (Parameterised Model № 3). В методе АМ1 (имеются параметры для атомов H, Li, Be, B, C, N, O, F, Al, Si, P, S, Cl, Zn, Ge, Br, Sn, I, Hg) скорректировано завышение остов–остовного отталкивания при RAB > 3Å, что сразу улучшило точность определения энергий образования органических молекул и энергий водородных связей. Метод PM3 отличается от АМ1 способом выбора и большим числом параметров, определенных по более чем 500 молекулам (параметризова-
ны атомы H, Li, Be, C, N, O, F, Na, Mg, Al, Si, P, S, Cl, Ca, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Se, Br, Cd, In, Sn, Sb, Te, I, Hg, Pb), призванными повысить точность расчета. Круг объектов метода PM3 уже включает соединения

30
переходных элементов. Оба метода обеспечивают возможность расчета энергии межмолекулярного взаимодействия, водородной связи и переходных состояний органических реакций с ошибкой менее 20 кДж/моль.
MNDO: параметры были определены по 32 молекулам. Пригоден только для расчетов молекул, состоящих из s– и p– элементов. Не описывает водородную связь и межмолекулярное взаимодействие, завышает барьеры активации. MNDO/d: позволяет рассчитывать d–элементы.
АМ1: параметры были определены по 100 молекулам (от 7 до 21 параметра на элемент). Как и MNDO, позволяет проводить расчеты молекул, состоящих из s– и p–элементов.
Метод АМ1 не описывает соединений с гипервалентными связями, завышает энергии диссоциации, активации молекул и барьера переноса протона.
Рисунок 18 показывает степень эффективности применения различных квантово–химических методов для оценки торсионных углов в органических молекулах:
Рис. 18.
РМ3: параметры оптимизированы по 657 молекулам (18 параметров на элемент). Пригоден для расчета гипервалентных соединений и молекул, содержащих d–элементы. Хорошо описывает геометрию молекул, водородную связь, теплоты образования. Завышает величину барьера переноса протона, энергию ван–дер–ваальсового взаимодействия. Непригоден для расчета потенциалов ионизации.
Полуэмпирические квантово–химические расчеты требуют адекватного выбора метода для постановки компьютерного эксперимента. Причем обилие методов затрудняет этот выбор для начинающего исследователя. Некоторые практические рекомендации по применению полуэмпирических методов собраны в таблицу 9.
15. Разделение σ– и π–электронов. π–электронное приближение
При квантово–химических исследованиях ненасыщенных и ароматических молекул, чаще всего являющихся плоскими, как правило, используют π– электронное приближение, состоящее в следующем. Для плоских молекул валентные АО можно разбить на две группы. Одна из них содержит орбитали, симметричные относительно отражения в плоскости молекулы (σ–АО), другая – орбитали, антисимметричные относительно такого отражения (π–АО). Рис. 19
31
иллюстрирует оба случая на примере этилена. σ–электроны имеют максимальную вероятность нахождения в плоско-
сти молекулы, т.е. локализованы близ нее; вероятность нахождения здесь π–электронов равна нулю. Из–за этого π– электроны слабее связаны с остовом молекулы, более подвижны, легче ионизируются и более активны во взаимодействиях.
Свойства ненасыщенных и ароматических систем – высокая реакционная способность, зависимость от донорных и акцепторных заместителей, спектральные характеристики и др. – определяются, в основном, именно электро-
нами, описываемыми π–орбиталями. Поэтому при решении уравнений Рутана для таких систем вводят π–электронное приближение (Хюккель, 1931 г.): валентные σ–АО считают неполяризованными и включают в атомный остов, а дви-
жение π–электронов рассматривают в потенциальном поле таких остовов. Волновая функция молекулы при этом представляется как произведение Ψ = Ψσ Ψπ, где Ψσ и Ψπ – нормированные антисимметричные по отношению к σ– и
π–электронам функции, соответственно. Их можно разложить по слейтеровским детерминантам, составленным только из σ– и только из π–МО. Волновая функция Ψσ одинакова как для основного, так и для возбужденных состояний, и
все изменения связываются с π–электронами. Существенно, что рассмотрение только π–электронов удовлетворяет вариационному принципу (Мак–Вини, 1954 г.; Лайкос и Парр, 1956 г.).
В результате размерность уравнений Рутана сильно сокращается: например, для этилена вместо 12 валентных электронов необходимо учитывать только 2 π–электрона.
Рис. 19. Молекулярные орбитали этилена. А - связывающие σ-орбитали: карта в плоскости молекулы; Б - пространственное представление связывающих σ-орбиталей: В, Г, Д, Е - карты и пространственное представление связывающих π - и разрыхляющих π*-орбиталей

32
16. Метод молекулярных орбиталей Хюккеля
Этот чрезвычайно простой метод, не использующий приближение самосогласованного поля, первоначально был предложен для углеводородов (Хюккель, 1931). Метод основан на нескольких сильных приближениях:
1) Принимаются π–электронное приближение; считают, что π–АО образуют ортонормированный базис, т.е. Sμν
= δμν.
2)Межэлектронными взаимодействиями (т.е. всеми двухэлектронными кулоновскими и обменными интегралами) пренебрегают, однако параметры метода учитывают их. Из–за этого решение уравнений метода не требует итераций и проводится в один шаг.
3)Матричные элементы оператора Фока оценивают на основании эмпирической информации и являются фиксированными:
hμμ = αμ ,
hμν = kβμν . (65)
αμ называется кулоновским интегралом (его не следует путать с двухэлектронными кулоновскими интегралами γAB),
и принимается равным потенциалу ионизации электрона на орбитали μ в свободном атоме αμ = Iμ. 4) Считают, что βμν = 0, если АО μ и ν не принадлежат связанным атомам.
После этих приближений уравнения Рутана имеют вид
∑ciμ (hμν −εiδμν ) = 0 |
(66) |
μ |
|
и называются уравнениями Хюккеля. Они решаются в один шаг и имеют ненулевые решения при равенстве нулю детерминанта
hμν – εiδμν = 0. |
(67) |
Полная энергия в методе Хюккеля есть просто сумма орбитальных энергий |
|
E = ∑niεi , |
(68) |
i |
|
где n = 0, 1 или 2 – число электронов на МО.
Рассмотрим решение уравнений Хюккеля на примере молекулы этилена С2Н4, имеющей 2 π–электрона (свяжем их с χ(р) АО атомов углерода, направленными перпендикулярно плоскости молекулы). МО имеет вид
φi = N(χ1 + χ2 ) . (69)
Величины необходимых интегралов следующие: αС = – 11.0 эВ, βСС = – 2.4 эВ. Детерминант (67) в рассматриваемом случае имеет вид
αc −ε |
βcc |
|
= |
|
x |
1 |
|
= 0 |
|
|
|
||||||
βcc |
αc −ε |
|
|
1 |
x |
|
||
|
|
|
|
|
(здесь произведена замена х = (αС – ε)/βСС). Раскрывая определитель, имеем х2 –1 = 0 и х = ± 1, откуда
ε1= αС +βСС , ε2 =αС – βСС.
В принятых обозначениях система уравнений имеет вид
с1 |
х + с2 = 0 |
с1 |
+ с2 х = 0, |
(70)
(71)
(72)