

33
Подставим х = ± 1 в (72). При х = –1 получим с1 = с2 . Используя далее условие нормировки волновой функции этилена с12 + с22 = 1, получаем: с1 =
= с2 = |
1 |
. Таким образом, одна из π–МО этилена имеет вид |
2 |
ϕ1 = |
1 |
(χ1 +χ2). |
(73) |
2 |
При х = 1 имеем с1 = – с2 и, повторяя рассуждения, получаем другую π–МО:
ϕ2 = |
1 |
(χ1 – χ2). |
(74) |
2 |
Обе π–МО этилена изображены на рис. 19.
Так как βСС < 0, то ε1 < ε2 причем, ε1 – ε2 = 2βСС. Это означает, что МО ϕ1 имеет более низкую энергию, т.е. более энергетически стабильна.
Полинг, Уэлланд, Стрейтвизер, Дьюар и другие предложили различные модификации метода Хюккеля, распространив его, в частности, на системы с гетероатомами в цикле. Модификация в основном касается способа выбора па-
раметров α и β.
16.1 Расширенный метод Хюккеля
Удачная модификация метода Хюккеля принадлежит Р. Хоффманну (1963). Он сохранил оригинальную схему Хюккеля, но включил в рассмотрение все валентные (а не только π) орбитали и явно учел интегралы перекрывания. В итоге уравнения метода, получившего название расширенный метод Хюккеля (РМХ), формально совпадают с уравнениями Рутана (21), однако содержание матричных элементов совершенно иное. Матричные элементы оператора Фока
Fμν ≡ hμν являются параметрами или оцениваются с помощью соотношений, включающих эти параметры. Наиболее часто используются следующие оценки:
|
hμμ = – Iμ. , |
|
|
hμν = 0.5K(hμμ + hνν). |
(75) |
В варианте Вольфсберга–Гельмгольца К = 1.75. Интегралы перекрывания вычисляются аналитически с ОСТ. |
||
Электронная энергия молекулы с закрытой оболочкой в РМХ является удвоенной суммой энергий занятых МО |
||
и описывается выражением |
|
|
E = 2∑(∑ci2μhμμ +∑∑ciμciν hμν ) , |
(76) |
|
i μ |
μ ν |
|
(члены, ответственные за межэлектронное и межъядерное взаимодействие, не учитываются). Выражение (76) дает хорошие относительные оценки энергии в рядах соединений с равномерным распределением электронов. Гетероатомы нарушают равномерность электронного распределения и РМХ в соединениях с гетероатомами часто непригоден.
17. Расчет свойств молекул
Энергия является важнейшей характеристикой молекул. Любой квантово–химический расчет основан на минимизации энергии системы N электронов и M ядер. Современные методы поиска минимума позволяют отыскать на поверхности потенциальной энергии минимум, отвечающий равновесной конфигурации молекулы с наименьшей энергией (или минимумы, если их несколько). Важную роль здесь играет выбор начальной геометрии и симметрии
34
молекулы: расчеты показывают, что при правильном учете симметрии из–за уменьшения количества варьируемых параметров время расчета может сократиться в несколько раз.
Дипольные и квадрупольные моменты молекул характеризуют распределение заряда по молекуле и определяют энергию межмолекулярного взаимодействия.
Дипольный момент
μ = ∫r[∑Zaδ |
|
e |
|
(r − Ra ) − ρмол(r)]dV = ∑Za |
|
e |
|
Ra −2∑∑Pμν ∫rχμ (r)χν (r)dV |
(77) |
|
|
|
|
||||||
|
|
|
|
|
|
|
|
μ ν |
|
( смол(r) – электронная плотность молекулы, Za и Ra – заряд и координата ядра) в концентрированном виде характеризует полярность молекулы. В химии принимают, что диполь направлен от центра тяжести положительных зарядов к центру тяжести отрицательных зарядов. Для электронейтральных молекул дипольный момент не зависит от выбора начала отсчета.
Абсолютные значения дипольных моментов могут быть определены методом молекулярных пучков, микроволновой спектроскопии, ИК–спектроскопией, измерением комплексной диэлектрической проницаемости как функции частоты и температуры. Сравнивая результаты расчета и измерений, можно оценить надежность проведенного экспериментального исследования. Однако только квантово–химический расчет позволяет определить точное направление (или знак) дипольного момента.
Результаты расчетов дипольных моментов некоторых молекул, используемых в технике или имеющих важное биохимическое значение, приведены в табл. 10. Можно заключить, что расчет, занимающий несколько минут, хорошо предсказывает экспериментальные значения дипольных моментов.
Табл. 10. Дипольные моменты некоторых молекул (×1030 Кл м)
Молекула |
Расчет |
Эксперимент |
|
(6–31G**) |
(измерения в газе или растворе) |
||
|
|||
Вода H2O |
+7.29 |
6.186 |
|
Цианамид CH2N2 |
+16.26 |
13.3 – 15.1 |
|
Формамид CH3NO |
+14.18 |
12.4(2) |
|
Мочевина CH4NO |
+17.06 |
15.2(1) |
|
L–аланин |
+41.44 |
41.0 |
|
|
|
|
|
Урацил C4H4N2O2 |
+16.22 |
13.9(1) |
|
пара–нитропиридин–N–оксид |
+1.00 |
2.3(1) |
|
C5H4N2O3 |
|||
|
|
||
|
|
|
Квадрупольный момент, характеризующий отклонение распределения заряда от сферического, играет особенно важную роль, если дипольный момент нейтральной молекулы равен нулю. Тогда именно он характеризует распределение электронной плотности и определяет электрическое поле вокруг молекулы. Существует несколько определений квадрупольного момента; наиболее часто используется следующее:
θαβ = |
1 |
∫[3rα rβ −r2δαβ ]ρ(r)dr, α, β = x, y, z |
(78) |
|
2 |
|
|
(δαβ – символ Кронекера). Из определения (78) видно, что квадрупольный момент есть симметричный тензор 2–го
ранга, причем сумма его диагональных элементов равна нулю. Для нейтральных молекул с нулевым дипольным мо-
ментом квадрупольный момент не зависит от выбора системы координат. Положительный знак θαβ указывает на
«вытянутое» вдоль оси z распределение заряда, отрицательный – на «сплющенное» вдоль оси z распределение.

35
Квадрупольный момент удобно выразить через так называемые вторые моменты электронного распределения
μαβ = ∞∫ rα rβ ρ(r)dr, |
α, β = x, y, z |
(79) |
−∞ |
|
|
где
θxx=μxx–(1/2)(μyy+μzz), θxy=(3/2)μxy.
Остальные компоненты θαβ получаются простой перестановкой индексов.
В таблице приведены вычисленные и измеренные компоненты квадрупольных моментов некоторых молекул. Из табл. 11 следует, что, хотя согласие хуже, чем в случае дипольных моментов, тем не менее простой неэмпирический расчет вполне удовлетворительно предсказывает их величины. Это обстоятельство позволяет, в частности, вести направленный поиск новых материалов, обладающих высокими нелинейными оптическими свойствами. Обуславливающие их электронные поляризуемости зависят от вторых и третьих моментов электронной плотности молекул в основном состоянии. Поэтому задача сводится к поиску веществ без центра симметрии, в которых взаимная ориентация молекул с большими вторыми и третьими моментами θxx, θуy, θzz будет обеспечивать высокие нелинейно–оптические характеристики.
|
Табл. 11. Квадрупольные моменты некоторых молекул (×1040 Кл м2) |
||
|
|
|
Эксперимент |
Молекула |
Компонента |
Расчет (6–31G**) |
|
|
|
|
+8.77(7) |
|
θxx |
+7.93 |
|
Вода Н2O |
θyy |
–7.59 |
–8.34(7) |
|
θzz |
–0.33 |
–0.34(1) |
|
|
|
–1.0(7) |
|
θxx |
–4.44 |
|
Формамид CH3NO |
θyy |
+12.58 |
–11.3(1) |
|
θzz |
–8.14 |
–10(3) |
|
|
|
+20.1(6) |
Ацетилен C2H2 |
θzz |
+23.23 |
|
|
|
|
+4.7 |
|
θxx |
+4.99 |
|
Этилен C2H4 |
θyy |
–11.04 |
–12.0 |
|
θzz |
+6.05 |
+7(1) |
|
|
|
–2.8(3) |
S–триазин C3H3N3 |
θzz |
+2.03 |
|
|
|
|
–3.1(9) |
|
θxx |
–1.58 |
|
Имидазол C3H4N2 |
θyy |
+17.43 |
+22(1) |
|
θzz |
–15.84 |
–20(2) |
|
|
|
|
Квадрупольные моменты даны относительно центров масс молекул. Координатные оси выбраны так, чтобы максимально учесть симметрию молекул: ось z направлена вдоль оси 2–го порядка для молекул с симметрией С2V
и перпендикулярно молекулярной плоскости для остальных молекул.
Молекулярный электростатический потенциал (МЭП) определяется электронной плотностью ρ(r) и зарядами ядер Za:
V ( r ) = − |
|
|
|
ρ ( r ) |
|
|
d Vi + ∑ |
|
|
Z α |
|
|
. |
(80) |
|
∫ |
|
r - r |
|
|
|
|
|
|
|
||||||
|
|
|
|
||||||||||||
|
|
|
|
α |
|
r - R |
α |
|
|
|
|
||||
|
|
|
|
i |
|
|
|
|
|
|
|
|
|
|
|
36
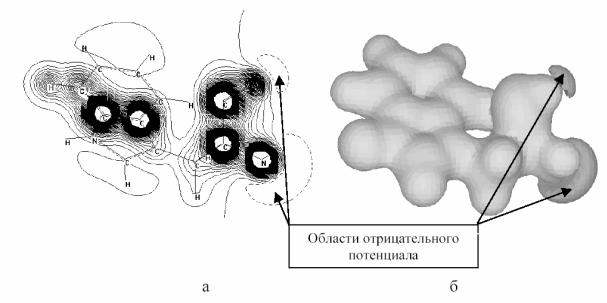
МЭП характеризует энергию электростатического взаимодействия между молекулярным (отрицательным и положительным) распределением заряда и единичным положительным бесконечно малым зарядом. От МЭП зависит сила Гельмана–Фейнмана, действующая на ядра молекулы при их отклонениях от положения равновесия, т.е. МЭП обуславливает равновесную конфигурацию молекулы. МЭП определяет также активные центры взаимодействующих молекул (рис. 20).
Рис. 20. Молекула L–изомера незаменимой аминокислоты TRP (триптофана). а – карта распределения молекулярного электростатического потенциала, б – пространственная картина МЭП.
18. Точность квантово–химических расчетов свойств молекул
Расчеты, дающие значения энергетических характеристик с ошибкой ± 10 кДж/моль, считаются выполненными с «химической точностью».
Неэмпирические методы. Ошибки всех неэмпирических квантово–химических методов возрастают при использовании «коротких» базисных наборов. Для органических молекул справедливы следующие оценки точности расчета методами ХФ или МР2 при базисе, не хуже чем 6–31G*:
•длины связей – 0.01–0.02 Å (для элементо– и металлоорганических соединений несколько хуже);
•валентные углы – ~ 1 %;
•электронная плотность –10%;
•энергии конформационных переходов (вращение и барьеры инверсии) – не хуже, чем 9 кДж/моль (желательно использование широких базисов);
•ХФ–частоты колебаний для большинства ковалентных связей систематически завышаются на 10–12 % из–за пренебрежения электронной корреляцией и ангармонизмом (очень низкие частоты колебаний имеют более высокие ошибки): масштабирующий множитель 0.89 ± 0.01 позволяет получить прекрасное согласие с экспериментом. Учет электронной корреляции по методу теории возмущений МР2 приводит к таким же результатам без поправки;
•энергии нулевых колебаний ~ 5 кДж /моль;
•энергии изодесмических реакций – 8–12 кДж /моль;
•присоединение/отщепление протона (в газовой фазе с включением аниона) – ~ 40 кДж /моль;
37
• реакции атомизации и гомолитического разрыва связи и барьеры активации реакций – 100–160 кДж /моль. Полуэмпирические методы. Полуэмпирические методы параметризуются таким образом, чтобы воспроизво-
дить те или иные свойства молекул. Поэтому расчет всех свойств на основании одного набора параметров надежным безусловно признан быть не может. Это общий и наиболее существенный недостаток полуэмпирической квантовой химии.
Для молекул с закрытыми оболочками методы AM1 и PM3 дают хорошие результаты для определения молекулярной структуры и теплоты образования. Средняя абсолютная ошибка для неводородных атомов в PM3 составляет 0.036 Å; она несколько больше в методе AM1. Ошибки в валентных углах равны 3–4 градуса. Это хуже, чем в неэмпирических расчетах даже низкого уровня, но время расчета несоизмеримо меньше. Ошибка PM3– расчета энергии образования органических молекул и переходных состояний органических реакций составляет менее 20 кДж /моль.
Тенденции в изменениях колебательных частот в рядах близких соединений могут быть выявлены с помощью полуэмпирических методов, однако отклонения (ошибки) не носят систематический характер и поэтому их не удается скорректировать.
Методы теории функционала плотности для небольших молекул обеспечивают точность, сравнимую с неэмпирическими расчетами с учетом корреляции электронов в очень широком базисе.