

Solid Support Oligosaccharide Synthesis
.pdf88 POLYMER-SUPPORTED SYNTHESIS OF OLIGOSACCHARIDES
4.4 SOLUBLE POLYMERS AS SUPPORTS
4.4.1 MPEG as Support
The use of polyethyleneglycol (PEG) derivatives as support for the solid-phase synthesis of oligosaccharides has been reported by Krepinsky et al. as early as 1991.10 In this report, these authors presented a synthesis of lactosamine from an immobilized glucosamine acceptor and peracetylated galactosyl donor in 70%. To our knowledge, this was the first example of a glycosidic bond formation using a polymer-supported acceptor and a trichloroacetimidate donor in solution. The support employed was polyethyleneglycol monomethyl ether (MPEG; average molecular weight = 5000). The acceptor was attached to the support via a succinate linkage and was cleaved by hydrazinolysis.
This strategy appears to be very attractive because of the possibility of completely solubilizing the support in most of the common solvents. From a chemical perspective, that property allows one to benefit from all the solvent conditions used in classical solution chemistry. This could prove to be very advantageous, especially to obtain stereoselective glycosylation without neighboring-group assistance. Moreover, isolation and purification of the polymer is easily achieved by precipitation usually in diethyl ether or methyl-tert-butyl ether (MTBE) and recrystallisation from ethanol. One major drawback of this type of support is its tendency to solidify at low temperature, thus limiting the variety of temperature conditions.
In a second publication,11 these authors improved their approach by designing a new linker (Scheme 4.15) synthesized from MPEG. The new support, MPEG-DOX (DOX = α,α′-dioxyxylyl), was prepared as MPEG-DOX-Cl in one ether synthesis step using α,α′-dichloro-p-xylene, or as MPEG-DOX-OH by hydrolysis from the latter. Compared with the succinate linker, the authors claimed a greater stability and the possibility of cleaving the target oligosaccharide by controlled hydrogenolysis as a free OH or as a p-tolylymethyl derivative. With this new linker strategy in hand, the synthesis of D-mannopentaose from MPEG-DOX-OH using donor 6 was performed and a reiterative sequence presented in Scheme 4.15.
The final compound was isolated after peracetylation; no trace of β-anomer was formed during the process. As in their first report, these authors also investigated the MPEG-DOX linkage of the first sugar residue via hydroxyl groups other than the anomeric one (Scheme 4.16).
Thus, synthesis of lactosamine 72 was reported using the new linker system and an 1-O-allyl glucosamine building block 68. Finally, the controlled hydrogenolysis concept was applied to the synthesis of lactose derivatives (Scheme 4.17).
Using 5% Pd black, 1 atm H2 in ethanol at r.t. for 48 h, p-tolylmethyl derivative 74 was selectively obtained. Under stronger conditions (10% Pd/C, 50% aqueous AcOH, 3 atm H2 at 50°C) MPEG-DOX was completely cleaved to release, after acetylation, the derivative 75. This selective cleavage was reproduced using different 2-O-protected or 2-O-glycosylated hexopyranoses.

Scheme 4.15 Synthesis of D-mannopentaose from MPEG-DOX-OH using donor 6.
89

90
Scheme 4.16 MPEG-DOX linkage of the first sugar residue via nonanomeric hydroxyl groups.
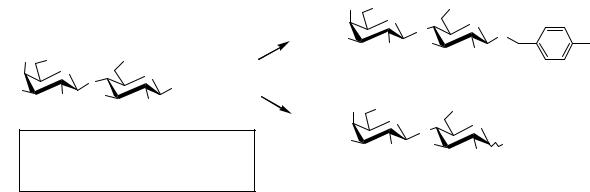
Scheme 4.17 Application of the controlled hydrogenolysis concept in lactose derivative synthesis.
91
92 POLYMER-SUPPORTED SYNTHESIS OF OLIGOSACCHARIDES
Studying the influence of the activator and using the MPEG-DOX-OH support (Scheme 4.18),12 the same group reported the synthesis of trisaccharide 79 using donors 76 and 78; both glycosylations were performed by action of 3 equiv of donor, in the presence of 0.1 equiv of dibutylboron triflate (DBBOTf) as activator and 4-Å molecular sieves at –45°C for 30 min.
Glycosylation of MPEG-DOX-OH with 76 was achieved in 95% yield. After deacetylation using DBU in methanol, glycosylation using disaccharide donor 78 was performed and resin-linked trisaccharide was obtained in 85% yield.
Dreef-Tromp et al. described the synthesis of O-methylated heparan sulfate–like oligomers up to the 12 mer on MPEG (average molecular weight = 5000) (Scheme 4.19).13 In a first stage, synthesis of the fully protected support bound oligosaccharides was achieved by reiterative delevulinoylation–glycosylation–capping elongation cycles. These syntheses started from resin-bound acceptor 80 and were based on the use of donor 81. The simple, base-sensitive succinate linker that was employed prompted the authors to use the levulinoyl moiety as temporary protecting group. Deprotection was easily performed under mildly basic conditions, which were compatible with the linker and the acetates used for capping, by action of hydrazinium acetate in pyridine. The glycosylations were optimized by performing the reaction at 10°C and by using 2.5 equiv of donor, 0.45 equiv of TMSOTf as activator, and 4 Å molecular sieves in dichloromethane. Couplings were found to work with more than 95% efficiency in this case. Unreacted hydroxyl groups were then capped by acetylation before performing the next elongation cycle. The cycles were completed twice only when the glycosylation efficiency proved to be less than 95%. Monitoring was effected by standard 1H NMR spectroscopy. The fully protected oligosaccharides were released from the support with concomitant removal of the acetyl protecting groups by action of lithium hydroperoxide. Subsequent debenzylation, O-sulfation, and purification of the target compounds were performed using standard techniques.
4.4.2 Low-Molecular-Weight MPEG as Support
Chan et al. reported the use of a low-molecular-weight poly(ethyleneglycol)- ω-monomethyl ether (MPEG; average molecular weight = 550, n = ~8–20).14 The advantage of this particular PEG derivative is the possibility of purification by conventional chromatographic methods and to characterize the product by using common NMR techniques. Here also the α-(1→2)-linked tetramannoside was chosen as the target using the abovementioned mannosyl donor 6. Glycosylation steps were performed using 2.0 equiv of donor in the presence of 0.8 equiv of TMSOTf and 4-Å molecular sieves. The reactions were performed in dichloromethane at room temperature. Deacetylations were achieved using potassium carbonate in wet methanol at r.t. for 4 h. Resin-linked tetrasaccharide was cleaved and debenzylated in one step using standard hydrogenation conditions. The tetrasaccharide was isolated after peracetylation in 10% overall yield.

Scheme 4.18 Glycosylation of MPEG-DOX-OH in trisaccharide synthesis.
93

94
Scheme 4.19 Synthesis of O-methylated heparan sulfate-like oligomers.
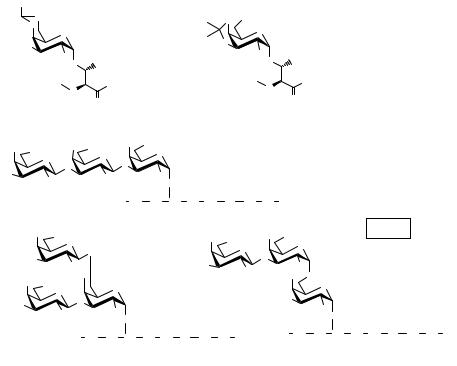
Scheme 4.20 Solid-phase synthesis of various octapeptides.
95
96 POLYMER-SUPPORTED SYNTHESIS OF OLIGOSACCHARIDES
4.5 OLIGOSACCHARIDE SYNTHESES ON PEPTIDES ATTACHED TO A SOLID SUPPORT
An attempt to synthesize an oligosaccharide chain by the trichloroacetimidate method using a glycosylated peptide as primer has been reported by Paulsen and Bock.18 The support employed here was a polyethylene glycol dimethylacrylamide copolymer (PEGA) derivatized with the well-known Rink amide as an acid-labile linker. This constitutes a standard system for solid-phase peptide synthesis. These authors performed the solid-phase synthesis of different octapeptides (Scheme 4.20) containing a threonine residue glycosylated with 2-azido-2-deoxy-α-D-galactose unsubstituted in position 3 or 6. To this end, buiding blocks 82 and 83 were employed.
The oligosaccharide chain was subsequently elongated by using different perbenzoylated trichloroacetimidate donors. Glycosylations were performed by using an excess of donor (8–10 mol equiv) in dichloromethane at low temperature (–15 to –30°C) and TMSOTf as catalyst. The reaction time used was 12–24 h. Reaction monitoring was performed by analytical cleavage of small resin samples. Different, linear (84) or branched (85, 86) trisaccharide-containing peptides were isolated after TFA-mediated cleavage from the support. Whereas the neighboring-group participation was successfully applied to 2-O-benzoylated donors or Teoc-protected 2-amino-2-deoxy donors with encouraging yields, the 2-azido-2-deoxy donors proved to give the expected α linkage, but only in modest yields. In the case of aminosugar donors, it is to be noticed that a change of support from PEGA to polyhipe proved to be favorable. Using this last support, these contributors attempted even a direct glycosylation of a pentapeptide containing a threonine residue with a free hydroxy group. All attempts made with different L-fucosyl and D-xylosyl donors gave no glycosylation product; however, under more forcing conditions, side reactions with tBu-protected glutamic acid led to α-glycosidically bound esters.
4.6 CONCLUSIONS AND OUTLOOK
O-Glycosyl trichloroacetimidates exhibited excellent glycosyl donor properties in polymer–supported oligosaccharide syntheses. In particular, linkage to the Merrifield resin as solid support turned out to be successful. As linkers, thioglycosides, 4-alkyloxybenzyl, and 4-acylaminobenzyl glycosides, and, above all, ring closing and cross-metathesis, offered efficient oligosaccharide synthesis on the resin and finally cleavage from the resin. Also, attachment of the first sugar to the resin via anomeric positions as either acceptor or donor was successfully probed.
Even controlled-pore glass (CPG) could be successfully employed as solid support with O-glycosyl trichloroacetimidates as glycosyl donors. Thus, limitations of solvents and reaction temperatures in the glycosylation step, as experienced with the Merrifield resin, are restricted to those observed in solution-phase synthesis. Therefore, regioand stereocontrol of the glycosylation reactions should be available from well-established solution-phase methodologies.
REFERENCES 97
Soluble polyethylene glycol (PEG) as polymer support was also successfully investigated. Precipitation after each reaction step in order to remove excesses of reagents and soluble byproducts seemed to work well. Another topic of great interest is direct glycosylation of solid support connected peptides in order to arrive finally at glycopeptides. O-Glycosyl trichloroacetimidates also proved successful in this endeavor.
These results demonstrate that O-glycosyl trichloroacetimidate–based oligosaccharide synthesis on solid support may eventually become a valuable alternative to solution-phase synthesis because useful experience is available for the selection of the polymer support and choice of the linker system and the glycosyl donor. Further standardization of the building blocks and the protective group pattern will finally provide the yields and the anomeric control in order to successfully plan automated syntheses of oligosaccharides also in a combinatorial manner.
REFERENCES
1.Rademann, J., and Schmidt, R. R., Tetrahedron Lett. 37, 3989–3990 (1996).
2.Rademann, J., and Schmidt, R. R., J. Org. Chem. 62, 3650–3653 (1997).
3.Rademann, J., Geyer, A., and Schmidt, R. R., Angew. Chem., Int. Ed. 37, 1241–1245 (1998).
4.Hunt, J. A., and Roush, W. R., J. Am. Chem. Soc. 118, 9998–9999 (1996).
5.Shimizu, H., Ito, Y., Kanie, O. and Ogawa, T., Bioorg. Med. Chem. Lett. 6, 2841–2845 (1996).
6.Fukase, K., Nakai, Y., Egusa, K., Porco, J. A., and Kusumoto, S., Synlett 1074–1075 (1999).
7.Knerr, L., and Schmidt, R. R., Synlett 1802–1804 (1999).
8.Andrade, R. B., Plante, O. J., Melean, L. G., and Seeberger, P. H., Org. Lett. 1, 1811–1814 (1999).
9.Doi, T., Sugiki, M., Yamada, H., Takahashi, T., and Porco, Jr., J. A., Tetrahedron Lett. 40, 2141–2144 (1999).
10.Douglas, S. P., Whitfield, D. M., and Krepinsky, J. J.,J. Am. Chem. Soc. 113, 5095–5097 (1991).
11.Douglas, S. P., Whitfield, D. M., and Krepinsky, J. J.,J. Am. Chem. Soc. 117, 2116–2117 (1995).
12.Wang, Z. G., Douglas, S. P., and Krepinsky, J. J., Tetrahedron Lett. 37, 6985–6988 (1996).
13.Dreef-Tromp, C. M., Willems, H. A. M., Westerduin, P., van Veelen, P., and van Boeckel, C. A. A., Bioorg. Med. Chem. Lett. 76, 1175–1178 (1997).
14.Jiang, L., Hartley, R. C., and Chan, T. H., J. Chem. Soc. Chem. Commun. 2193–2194 (1996).
15.Heckel, A., Mross, E., Jung, K.-H., Rademann, J., and Schmidt, R. R.,Synlett 171–173 (1998).
16.Adinolfi, M., Barone, G., De Napoli, L., Iadonisi, A., and Piccialli, G.,Tetrahedron Lett. 39, 1953–1956 (1998).