

Solid Support Oligosaccharide Synthesis
.pdf168
*
x
Scheme 8.1 Synthesis of a sialyl-Lewis decoupling NMR spectroscopy.
*
*
13
tetrasaccharide employing C-enriched protecting groups for the quantitative reaction monitoring using gated
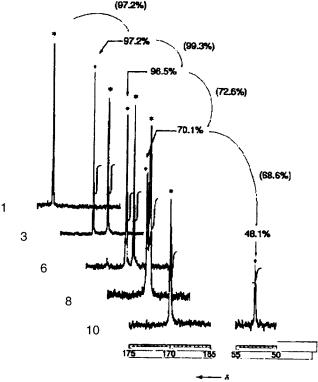
8.3 NMR SPECTROSCOPIC METHODS 169
Figure 8.2 Parts of the 13C spectra of the functionalized resins 1, 3, 6, 8, and 10. Yields were calculated from the signal intensities.17
it requires the use of 13C-enriched protecting groups. Furthermore, no information about the anomeric composition of the generated oligosaccharide was obtained.
For real on-bead analysis that goes beyond mere monitoring, it is desirable to obtain structural information from oneand two-dimensional proton NMR experiments. Purity, connectivity, and the presence or absence of functional groups can ideally be assessed without requiring isotopic labeling. High-resolution magic-angle spinning NMR (HR-MAS) techniques have been developed,18 which allow high-resolution 1H spectra to be obtained from resin-bound molecules. The major difficulty in using 1H spectroscopy to analyze a resin-bound molecule is the intrinsic linewidth.19 Solid-state NMR techniques are not sufficient to reduce the proton linewidths to the point where J couplings are observable. HR-MAS employs a solvent to swell the resin, thereby imparting mobility, which allows averaging of dipolar and chemical shift anisotropy (CSA) effects that are responsible for the large linewidths in solid-state spectra. This produces a spectrum (Fig. 8.3a) that is still broad by solution standards, but that definitely begins to show structural features (based on chemical shift). By spinning the slurry at the magic angle at rates of 1–2 kHz, residual dipolar and CSA effects, as well as magnetic susceptibility differences (due to the

170 TOOLS FOR “ON-BEAD” MONITORING
Figure 8.3 Illustration of HR-MAS techniques applied to a resin-bound trisaccharide: (a) static 1H spectrum of the solvent swollen sample; (b) 1H spectrum with magic-angle spinning at 3.5 kHz; (c) 1H spectrum with MAS and spin echo pulse sequence.
heterogeneous nature of the sample) are significantly reduced, narrowing 1H linewidths to the same order as 1H–1H J couplings (Fig. 8.3b). Employing a spin-echo pulse sequence (Fig. 8.3c) allows the discrimination of signals based on T2 relaxation rates, which results in a spectrum where most of the resin resonances have been significantly attenuated and the 1H–1H J couplings of the resin-bound moiety are visible.
In addition to well-resolved one-dimensional (1D) 1H and 13C spectra, which are usually sufficient for monitoring synthetic steps, HR-MAS techniques can be applied to two-dimensional (2D) homonuclear and heteronuclear experiments, which allow a wealth of structural information to be obtained. 1H,13C HMQC (heteronuclear multiple quantum coherence) spectra are particularly useful in the analysis of solid support-bound oligosaccharides, since the anomeric protons exhibit characteristic resonances. Such a spectrum of a polymer-bound trisaccharide glycal is shown in Figure 8.4.

8.3 NMR SPECTROSCOPIC METHODS 171
Figure 8.4 Example of an HR-MAS HMQC spectrum of a resin-bound trisaccharide glycal.20
We have characterized a resin-bound pentasaccharide by HR-MAS techniques. A comparison of the solution spectrum of the resin-cleaved pentasaccharide with the HR-MAS spectrum of the resin-bound pentasaccharide is shown in Figure 8.5. It is immediately obvious that the HR-MAS technique provides data of a quality similar to that of the solution technique, but in both cases, only four of the five anomeric protons are visible. However, a 2D homonuclear total correlation spectroscopy (TOCSY) spectrum (Fig. 8.6) of the resin-bound pentasaccharide allowed us to clearly observe the overlapped anomeric protons (demonstrating a resolution of 4.4 Hz).
The power of the HR-MAS method for on-resin analysis has been further underscored in the development of new linkers. Without this method, only indirect analytical data after removal from the resin was available. Direct assessment of the resin-bound linker greatly facilitated the introduction of a 4,5-dibromo octane-1,8-diol linker that was converted into an octane-1,8-diol linker cleavable by olefin metathesis at the end of the synthesis.6 The disappearance and reappearance of the olefinic protons as well as the growing oligosaccharide chain was clearly visible in the 1H spectrum (Fig. 8.7).7
Most of the methodology used so far in solid-phase oligosaccharide synthesis was developed before the analytical techniques outlined above had been adopted. Now that a closer insight into the reactions has become available, one may expect exciting

172 TOOLS FOR “ON-BEAD” MONITORING
Figure 8.5 Comparison of 1H,13C HMQC spectra obtained on resin-cleaved and resin-bound samples of a pentasaccharide.
Figure 8.6 1H,1H TOCSY spectrum of resin-bound pentasaccharide.

173
7
Figure 8.7 HR-MAS spectra of resin-bound intermediates in the solid-phase synthesis of a trisaccharide using thiodonors. Traces a and b have been recorded using spin echo pulse sequences to suppress resonances of the resin. Traces c and d have been recorded using a standard proton experiment.
174 TOOLS FOR “ON-BEAD” MONITORING
progress in future studies in this field that will facilitate the development of standardized procedures for the construction of an automated oligosaccharide synthesizer.
REFERENCES
1.(a) Rodebaugh, R., Joshi, S., Fraser-Reid, B., and Geysen, M. H., J. Org. Chem. 62, 5660–5661 (1997); (b) Rademann, J., Schmidt, and R. R., Tetrahedron Lett. 37, 3989–3990 (1996); (c) Rademann, J., and Schmidt, R. R., J. Org. Chem. 62, 3650–3653 (1997); (d) Rademann, J., Geyer, A., and Schmidt, R. R., Angew. Chem., Int. Ed. 37, 1241–1245 (1998).
2.(a) Yang, B., Acct. Chem. Res. 31, 621–630 (1998); (b) Shapiro, M. J., Lin, M., and Yan, B., in A Practical Guide to Combinatorial Chemistry, Czarnik, A. W., and De Witt, S.
H.(Eds.), ACS Professional Reference Book, Washington, DC, 1997, pp. 123–151.
3.Pivonka, D. E., J. Combinatorial Chem. 2, 33–38 (2000).
4.Li, W., and Yang, B., J. Org, Chem. 63, 4092–4097 (1998).
5.Rahman, S. S., Busby, D. J., and Lee, D. C., J. Org. Chem. 63, 6196–6199 (1998).
6.Andrade, R. B., Plante, O. J., Melean, L. G., and Seeberger, P. H., Org. Lett. 1, 1811–1814 (1999).
7.Melean, L. G., Haase, W.-C., and Seeberger, P. H., Tetrahedron Lett. 41, 4329–4333 (2000).
8.Nicolaou, K. C., Mitchell, H. J., Fylaktakidou, K. C., Suzuki, H., and Rodríguez, R. M.,
Angew. Chem., Int. Ed. 39, 1089–1093 (2000).
9.Hummel, G., and Hindsgaul, O., Angew. Chem., Int. Ed. 38, 1782–1784 (1999).
10.Rademann, J., and Schmidt, R. R., Carbohydr. Res. 269, 217–225 (1995).
11.Smith, B. C., Fundamentals of Fourier Transform Infrared Spectroscopy, CRC Press, Boca Raton, FL, 1996.
12.Chan, T. Y., Chen, R., Sofia, M. J., Smith, B. C., and Glennon, D.,Tetrahedron Lett. 38, 2821–2824 (1997).
13.Knerr, L., and Schmidt, R. R., Synlett 1802–1804 (1999).
14.Hanessian, S., and Huynh, H. K., Synlett 102–104 (1999).
15.Giralt, E., Rizo, J., and Pedroso, E., Tetrahedron 40, 4141–4154 (1984).
16.Shoolery, J. N., Prog. Nucl. Magn. Reson. Spectrosc. 11, 79–93 (1977).
17.Kanemitsu, T., Kanie, O., and Wong, C.-H., Angew. Chem., Int. Ed. 37, 3418–3420 (1998).
18.(a) Keifer, P. A., Baltusis, L., Rice, D. M., Tymiac, A. A., and Shoolery, J. N.,J. Magn. Res. A 119, 65–75 (1996); (b) Sarkar, S. S., Gargipati, R. S., Adams, J. L., and Keifer,
P.A., J. Am. Chem. Soc. 118, 2305–2306 (1996); (c) Fitch, W. L., Detre, G., Holmes,
C.P., Shoolery, J. N., and Keifer, P. A., J. Org. Chem. 59, 7955–7956 (1994); (d) Anderson, R. C., Jarema, M. A., Shapiro, M. J., Stokes, J. P., and Ziliox, M., J. Org. Chem., 60, 2650–2651 (1995).
19.Lippens, G., Bourdonneau, M., Dhalluin, C., Warrass, R., Richert, T., Seetharaman, C., Boutillon, C., and Piotto, M., Curr. Org. Chem. 3, 147–169 (1999).
20.Seeberger, P. H., Beebe, X., Sukenick, G. D., Pochapsky, S., and Danishefsky, S. J.,
Angew. Chem., Int. Ed. 36, 491–493 (1997).
Solid Support Oligosaccharide Synthesis and Combinatorial Carbohydrate Libraries. Edited by Peter H. Seeberger Copyright © 2001 John Wiley & Sons, Inc.
ISBNs: 0-471-37828-3 (Hardback); 0-471-22044-2 (Electronic)
9Polyethyleneglycol ω-Monomethylether (MPEG)-Supported Solution-Phase Synthesis of Oligosaccharides
JIRI J. KREPINSKY and STEPHEN P. DOUGLAS
Department of Medical Genetics and Microbiology, University of Toronto, Toronto, Ontario, Canada
9.1 INTRODUCTION
It is now accepted that carbohydrates, including oligosaccharides, usually in combination with lipids and proteins, display diverse biological activities, and that the importance of most of them is not fully understood yet.1 It is also agreed to that to improve this understanding, reasonably large quantities of pure oligosaccharides are needed.2,3 These oligosaccharides can be made chemically, but this is a time-consuming process because chromatography must be used in most cases. Polymer-supported methods decrease considerably the time spent on purification. The polymer support is usually solid, therefore it is called solid-state synthesis. Normally quantities of oligosaccharides assembled on solid support are quite small, perhaps because it is difficult to handle large-scale two-phase reactions, and because some solid supports are relatively fragile. It appears to be easier to make larger quantities when all reaction components are in solution; that is, the polymer support should be dissolved in the reaction solvent at the time of its use as a part of a reactant. Reactions usually proceed faster when all reaction components are dissolved (unless a catalyst participates in the reaction). After the reaction is completed, the polymer with the oligosaccharide product is then rendered solid, to remove soluble impurities as with solid supports. The most important among the soluble but precipitable polymers is polyethyleneglycol, mostly its ω-monomethylether (MPEG). MPEG is particularly useful for syntheses of combinatorial oligosaccharide libraries, in which case the polymer support is essential to purify the desired intermediates or products as groups. PEG support has been used to synthesize larger quantities of short oligopeptides and oligonucleotides,4 and as a general aid in organic synthesis.5
175
176 (MPEG)-SUPPORTED SOLUTION-PHASE SYNTHESIS OF OLIGOSACCHARIDES
The solid-phase synthesis of oligosaccharides started in the early 1970s in Schuerch’s group6,7 with unsatisfactory results; among the problems were decreased reaction rate and lower yields compared to solution strategies, incomplete coupling, and poor anomeric specificity. These problems seemed to be attributed to the solid phase rather than its incompatibility with certain aspects of Koenigs–Knorr chemistry. It was suggested at that time that the use of a soluble polymer could improve the results,8 but it took another 20 years until polyethyleneglycol as soluble support was examined.9 It was also shown at this time9 that the Koenigs–Knorr reaction (promoted by silver triflate) and trichloroacetimidate chemistry gave comparable results on MPEG, apparently confirming the assumption that the solid support (two-phase system) caused the failure of glycosylations. This conclusion became disputed when more data on two-phase synthesis were collected, and now it appears10 that there are no significant differences between glycosylations performed in solution without a polymer support, with MPEG as a soluble polymer support, or using a solid polymer support,11 although some variations were observed.12 Insoluble catalysts are much more likely to be effective in a solution, that is, with a soluble support, as exemplified by the removal of boronate diester from 4,6-positions of galactose by Amberlite IRA-743.13
The difference between the solid-state (two-phase) synthesis and solution-phase (one-phase) synthesis pertains to the reaction stage. In the isolation stage, the products are supported by a solid polymeric support. Both one-phase and two-phase designs are depicted in Scheme 9.1; oligosaccharide chains can be extended (A) from the reducing end to the nonreducing terminus (using polymer-supported acceptors), as well as (B) from the nonreducing end to the reducing terminus (using polymer-supported donors) (Scheme 2). Both strategies (A and B) share problems attributable to incomplete glycosylations and/or donor decomposition. Problems caused by incomplete glycosylation tend to increase as the oligomer becomes longer; assuming that a small portion (probably ~ 3–5%) of polymer-supported monosaccharide M1 acceptor fails to be glycosylated to a disaccharide D1, the disaccharide D1 in the subsequent step is extended to a trisaccharide T1, but a portion of the previously unglycosylated monosaccharide M1 gives disaccharide D2. These second round glycosylations are again incomplete. After the third glycosylation the following compounds will be polymer-bound in addition to the desired tetrasaccharide: the monosaccharide M1; disaccharides D1, D2, and D3; and trisaccharides T1 and T2. After each additional glycosylation the mixture becomes even more complicated, but all impurities originating from the donors are absent. The mixture is detached from the polymer support, and the desired product must be separated from the truncated sequences (= failures). To minimize formation of truncated sequences, excess glycosylation reactants are recommended to be applied in portions (Scheme 9.2), as is practiced to obtain “difficult sequences” in polymer-supported oligopeptide syntheses. Incomplete anomeric specificity causes further complications for oligosaccharide syntheses.
Starting the synthesis from the nonreducing terminus, that is, using a polymer-bound donor, presents a similar problem of truncated sequences. Donors are

Scheme 9.1 Comparison of one-phase and two-phase polymer-supported synthesis of oligosaccharides. Continued next page.
177