

Solid Support Oligosaccharide Synthesis
.pdf68
Scheme 4.1 A thiol functionalized resin.
4.2 POLYSTYRENE-BASED SUPPORTS 69
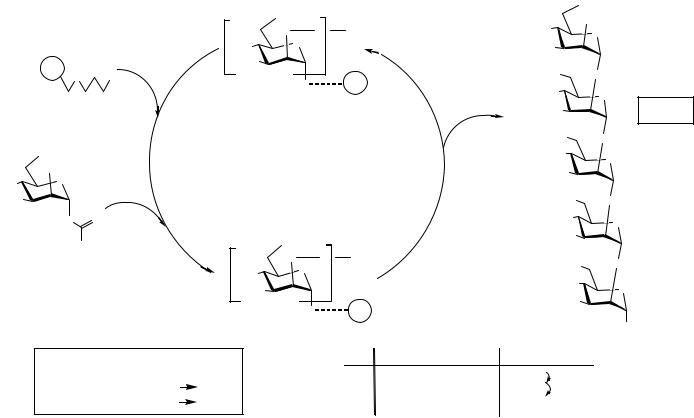
Among the different supports, resins 2 and 3, simply derived from Merrifield resin by O-alkylation using the corresponding protected thiol compounds or S-alkylation of symmetric 1,3-propanedithiol for 2, proved to be the most efficient in terms of reaction yields and chemical stability. These two resins turned out to be very good glycosyl acceptors, thus allowing a very efficient introduction of the first sugar residue. On the basis of this linker strategy, oligosaccharide synthesis was performed, first using 2 as support. Scheme 4.2 depicts the synthesis of glucose (1→6) oligomer using donor 5.
The anomeric anchoring of the glycosidic chain as a thioether allowed the final liberation of the oligosaccharide part using classical glycosylation conditions for thioether glycosyl donors. Dimethyl(methylthio)sulfonium salts were used as the thiophilic reagents, buffered with Hünig’s base in dichloromethane, and methanol as the glycosyl acceptor. Released compounds can thereafter be purified as their 1-O-methyl glycosides. This synthesis was completed until the stage of the pentamer. This new linker system allowed the analytical cleavage of small resin samples and thus the monitoring of reactions using MALDI-TOF (matrix-assisted laser desorption ionization–time of flight) mass spectrometry. The initial loading of 0.4–0.6 mmol/g (S elemental analysis of 2) proved to be too high to allow solid-phase reactions, and most of all glycosylations, to reach completion. The use of resin 3 was found to be more advantageous because of the greater chemical stability of the ether linkage during different reaction conditions. Moreover, lower and thus more practical loading was easily obtained (0.1–0.3 mmol/g, S elemental analysis). By taking advantage of these two improvements, more complex synthetic targets were envisaged. The synthesis of an α-(1→2)-linked pentamannoside 7 was then accomplished2 (Scheme 4.3) from resin 3 (0.2 mmol/g) using mannosyl donor 6.
This synthesis demonstrated that the neighboring-group participation effect on the stereoselectivity of glycosylation reactions can be extended to solid-phase processes. In this case, milder and more practical cleavage conditions than previously discussed were established. The use of N-bromosuccinimide as the thiophilic reagent in acetone/water or tetrahydrofuran/methanol permitted the release of oligosaccharides in form of lactols or 1-O-Me glycosides, respectively. The tetrasaccharide derivative was isolated in 34% yield from thiol resin 3 (80% yield per step).
Using the same linker system, the synthesis of disaccharide 10 was reported (Scheme 4.4). This last work nicely illustrated the application of the use of the anomeric effect for the synthesis of a 1,2-cis-glycosidic linkage in the absence of possible neighboring-group effects. The target molecule was prepared using fucosyl donor 9 for the glycosylation of attached acceptor 8. In solution, this type of glycosylation requires the combined use of diethyl ether as solvent and low temperatures for good stereocontrol. Unfortunately, the Merrifield resin derivative used here does not swell in diethyl ether alone. However, the judicious use of a 1/1 mixture of dichloromethane and 1,4-dioxane at –25°C afforded the target disaccharide in 54% overall yield as pure α-anomer.
With this solid-phase methodology in hand, the synthesis of branched oligosaccharides could be investigated. This led to the preparation of pentasaccharide 15 related to N-glycan chains (Scheme 4.5).3

70
Scheme 4.2 Synthesis of glucose (1→6) oligomer using donor 5.
71
Scheme 4.3 Synthesis of an α-(1→2)-linked pentamannoside 7 from resin 3.

72
Scheme 4.4 Synthesis of disaccharide 10.
4.2 POLYSTYRENE-BASED SUPPORTS 73
Starting from thiol resin 3, the first glycosylation using mannosyl donor 11 furnished resin 12. After deacetylation, acceptor resin 13 was used as the starting material for a glycosylation–deprotection–glycosylation sequence in which two glycosidic bonds were formed simultaneously during each glycosylation step. The target pentasaccharide was obtained in 20% overall yield (82% per step). The strategy used for this work was based on the use of neighboring-group participation to ensure stereoselective 1,2-trans-glycosylation using the previously described mannosyl donor 6 and Teoc-protected glucosamine donor 14. The pentasaccharide 15 is one of the most complex oligosaccharides synthesized on a solid support using a very elaborate and efficient synthetic approach.
4.2.2 Sulfonate Linkers
Another sulfur-containing linker was described by Hunt and Roush in 1996 (Scheme 4.6).4
The sulfonyl chloride resin 16 was obtained in three steps (70% overall) from Merrifield resin. The loading was determined by gravimetric analysis to be 0.65 mmol/g. This resin allowed for the attachment of the first sugar residue 17 using classical sulfonylation conditions. It should be noticed that in this case the first sugar residue is linked to the solid support via the C6 hydroxyl, thus giving access to 6-deoxy compounds. The authors described the use of acetyl esters and triethylsilyl ethers as temporary protecting groups for the synthesis of trisaccharide 22. The first glycosylation was accomplished using trichloroacetimidate donor 19 at low temperature. This step was performed two times in similar conditions to ensure a complete reaction. The target compound was released as a 6-deoxy-6-iodo derivative by nucleophilic substitution at the sulfur atom using NaI in 2-butanone at 65°C for 20 h. Subsequent radical reduction led to the corresponding 6-deoxy derivative. Analytical cleavages were performed at each stage of the oligosaccharide chain elongation. Thus disaccharide 21 was recovered, after on-bead acetylation of the corresponding acceptors and cleavage in 87% yield from glycal 18. The target trisaccharide was prepared in 67% overall yield (nine steps from attached glycal 18). It is remarkable that a glycosylation using donor 19 was accomplished without anchimeric assistance from a neighboring group. Use of low temperatures ensured excellent stereoselectivity control since the authors reported the isolation of only trace amounts of α-linked disaccharide; this corresponds to an improvement compared to the corresponding glycosylation in solution.
4.2.3 Substituted Benzyl Ether Linkers
A major contribution to the use of glycosyl trichloroacetimidates in the solid-phase synthesis of oligosaccharides came from the group of T. Ogawa (Scheme 4.7).5
These authors developed the use of a 4-alkyloxybenzyl ether–type linker attached to the Merrifield resin via an ester function that allowed the cleavage of the oligosaccharides under two different conditions. Using the reactivity of the activated

74
Scheme 4.5 Preparation of pentasaccharide 15.

75
Scheme 4.6 A sulfonate linker.

76
Scheme 4.7 Cleavage of attached oligosaccharides.
4.2 POLYSTYRENE-BASED SUPPORTS 77
benzylic position, they described the cleavage of attached oligosaccharides under mildly acidic conditions (TrBF4 in CH2Cl2 at room temperature) as a free hydroxyl derivative. The alternative approach is to use a simple basic transesterification as cleavage reaction (NaOMe in MeOH) in order to release the desired compound as its 4-alkyloxybenzyl ether derivative. To deal with an acidand base-sensitive linker, these authors had to develop a new protecting group pattern for the synthesis of linear polylactosamine oligosaccharides. The choice of the levulinoyl group as temporary protecting group permitted mild deprotection reactions using hydrazinium acetate, conditions fully compatible with the linker system. Starting from Merrifield resin (0.66 mmol/g) the first lactose unit was introduced using an ester synthesis step to arrive at resin 23. Extension of the chain was achieved using the lactosamine donor 24. The choice of phthaloyl as amino protecting group ensured the formation of the pure β linkage. The glycosylation steps (1.5 equiv donor, 0.2 equiv TMSOTf, CH2Cl2, –78°C, 2 h) were performed twice at each stage. Using a modified linker system, basic cleavage after the second elongation step furnished methyl ester 25 in 56% overall yield. Extension to the octasaccharide and subsequent acidic cleavage permitted the isolation of octasaccharide 26 in excellent 42% yield. Although mentioned by the authors, no oxidative cleavage using the well-known 4-alkyloxybenzyl reactivity of the linker was reported. There was no mention of analytical reaction monitoring.
A publication by Kusumoto et al.6 added two more perspectives for the solid-phase synthesis of oligosaccharides using trichloroacetimidate donors on polystyrenederived supports (Scheme 4.8). First, these authors described the use of a new 4-acylaminobenzyl ether linker that should be more acid-stable than the corresponding 4-alkyloxybenzyl derivative and could be easily cleaved under mild oxidative conditions (DDQ in CH2Cl2/H2O:20/1). To test this concept, the acid 27 was prepared in solution using classical techniques. This compound was subsequently attached to an aminomethyl polystyrene support (ArgoPore) using standard conditions for peptide bond formation and allowing the introduction of the linker and of the first sugar residue in one step.
The second innovation brought by these authors is the use of a highly crosslinked macroporous poly(styrene–divinylbenzene) support. The great advantage of this support is its solvent tolerance notably of diethyl ether and methanol, which could considerably extend the interest of trichloroacetimidate donors for the synthesis of oligosaccharides on this type of support. These contributors also suggest a greater accessibilty from the reagents to the reactive sites on the solid support due to the larger pore size (90 Å for the dry resin). Starting from partially protected resin-attached glucose 28, they tried to take advantage of the new support in order to investigate the possibility of α-selective glycosylations. Therefore, they used glucosyl donor 29 in diethyl ether at room temperature. By optimizing the support loading in acceptor and the glycosylation conditions, an isolated yield of 82% of (1→6)-linked disaccharide 31 as α/β mixture (α/β: 65/35) was obtained after cleavage. The trichloroacetimidate donor proved to give a good combination of yield and selectivity compared to other donors. Subsequent deprotection of the Teoc group [1 M NaOMe in methanol at room temperature (r.t.)] and glycosylation under the conditions described above gave after cleavage the corresponding trisaccharide 33 in 70% yield from 28.