

Solid-Phase Synthesis and Combinatorial Technologies
.pdf2.2 OLIGONUCLEOTIDES 63
that it links the growing ON through the heterocyclic base and not through the sugar. This allows a flexible strategy for incorporation of modified sugars in the terminal position. Three general supports able to support any starting ON or ODN (oligodeoxyribonucleotides) were recently reported (99–101). The majority of the cleavage procedures for automated ON synthesis use hot aqueous condition bases such as ammonium hydroxide, potassium carbonate, or other bases. A recent paper (102) reported that cleavage could be effected with gaseous ammonia under pressure, thus allowing easier work-up and washing procedures and giving better purity of target ONs.
The most popular protecting group strategy uses trityl-based protecting groups 2.37–2.40, which are reported in Fig. 2.15. The relative stability of ONs to acidic conditions means that mild acidic deprotection of the trityl-based protecting groups can be employed during the sequence, but stability during the coupling step is also required. The trityl 2.37 and trimethoxytrityl 2.40 groups are not suited to
|
R1 |
|
|
|
|
|
|
R2 |
|
|
|
|
|
2.37 |
R=R1=R2=H |
Trityl (Tt) |
|
|
|
2.38 |
R=OMe, R1=R2=H |
MeOTrityl (MMTt) |
|
|
O |
2.39 |
R=R1=OMe, R2=H |
DiMeOTrityl (DMTt) |
|
R |
|
2.40 |
R=R1=R2=OMe |
TriMeOTrityl (TMTt) |
|
|
HO |
O |
|
|
|
|
|
|
|
|
|
|
|
NB(P) |
|
|
|
R1 |
O |
|
|
|
|
|
O |
|
|
O2N |
NO2 |
|
O |
|
|
|
|
|
HO |
O |
|
|
|
|
|
|
2.42 |
R1=DNPEoc |
|
|
|
NB(P) |
|
||
|
|
|
|
|
|
2.41R1=Fmoc
R2
R2
|
|
S |
|
|
R1 |
|
2.43 |
R1=H, R2=H |
|
|
|
O |
||
|
|
2.44 |
R1=Ph(condensed), R2=H |
|
R1 |
|
O |
||
|
2.45 |
R1=Ph(condensed), R2=Me |
||
|
|
|
||
O |
O |
|
|
|
|
|
HO |
O |
|
|
|
|
|
|
NB(P)
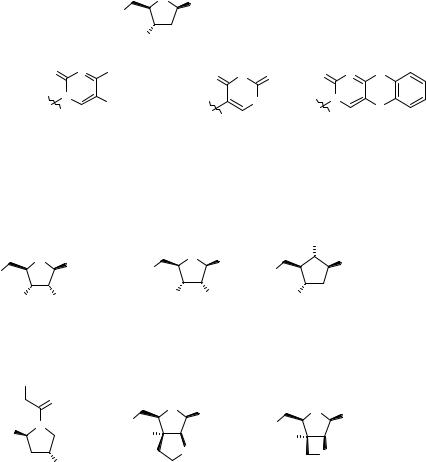
Figure 2.15 5′-protecting groups 2.37–2.45 for oligonucleotide SPS.

64 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
SP ON synthesis, being too stable and unstable, respectively. However, the monomethoxytrityl (MMt) 2.38 and the dimethoxytrityl (DMTt) 2.39 protecting groups possess the desired stability characteristics (103) and are used as 5′-protecting groups. In particular, DMTt has rapidly become the 5′-protecting group of choice (104) as it is easily cleaved under a variety acidic conditions, including 80% aqueous AcOH, BF3 Et2O, and ZnBr in various solvents or solvent mixtures (79). The base-labile 5′-protecting groups sometimes encountered are the carbonates, among them the Fmoc 2.41 (105, 106) and the DNPEoc (dinitro phenethyl carbonyl) 2.42 (107, 108) derivatives. The latter is cleaved by a TEA-promoted β-elimination. Finally, Hg-labile S-containing 5′-protecting groups such as 2.43–2.45 (Fig. 2.15) (109, 110) are more stable to basic hydrolysis but are readily cleaved with mercury salts. For example, 2.45 is cleaved by Hg(ClO4)2 in a few minutes.
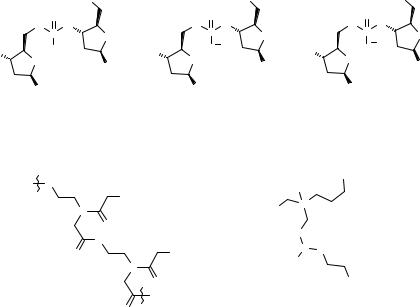
A recent review (79) gives details of other 5′-protecting groups, 2′-protecting groups for RNA-based ONs, and the use of specific protecting groups for nucleobases. Among the more recent developments, N-pent-4-enoyl (NPT) amide has been advocated as a universal nucleobase protecting group (111) applied to the building blocks 2.46–2.51 (Fig. 2.16), which are suitable for ON SPS. Protected NPT A, C, and G nucleosides are stable and can be employed in both amidite and phosphonate chemistries for the preparation of standard ONs or modified oligonucleotide analogs. Cleavage is carried out under either oxidative (iodine-base) or basic aqueous/anhydrous conditions. Other recent reports highlight the use of phthaloyl protections for the amine
|
O |
NB(NPT) |
|
DMTt |
O NB(NPT) |
|
HO |
|
O |
O |
|||
|
|
HO |
||||
O |
|
CPG |
L |
O |
O |
|
|
|
|||||
P |
NR2 |
|
O |
O |
O P H |
|
P1 O |
|
|
L = LINKER |
|
O |
|
NPT-phosphoramidites |
NB(NPT) |
NPT-phosphonates |
||||
|
||||||
NBs(NPT):
|
|
O |
|
O |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
NH |
|
NH |
N |
|
|
|
|
||
|
|
|
|
|
|
|
N |
O |
|||
|
N |
|
|
|
|
N |
|
||||
|
N |
|
N |
|
|
|
|
||||
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
N |
N |
|||
|
|
|
|
|
|
|
H |
||||
|
N |
N |
|
N O |
|
H |
|||||
|
H |
|
|
|
|
|
|
||||
2.46 |
|
|
|
2.47 |
H |
2.48 |
|
|
|
|
|
A-NPT (phosphoramidite) |
C-NPT (phosphoramidite) |
G-NPT (phosphoramidite) |
|||||||||
2.49 |
A-NPT (phosphonate) |
2.50 |
C-NPT (phosphonate) |
2.51 |
G-NPT (phosphonate) |
||||||
Figure 2.16 Nucleobase NPT-protected building blocks 2.46–2.51 and SP constructs for oligonucleotide SPS.

2.2 OLIGONUCLEOTIDES 65
functions of the nucleobases (112) and of photolabile protections for both sugars and nucleobases (113, 114) in automated RNA and DNA synthesis.
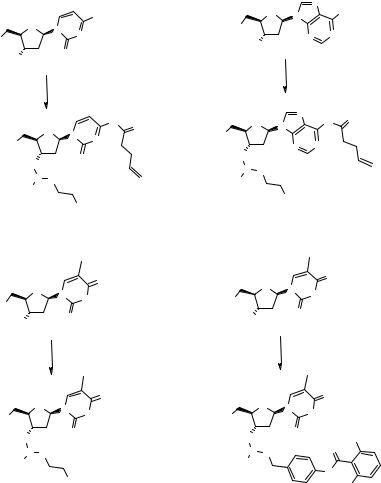
The chemistry of the phosphoramidite coupling has been thoroughly studied, and the use of 1H-tetrazole (115) as an activating agent for the reaction (Fig. 2.17) with support-bound 2.52 allows the SPS of long ON sequences with high yields (116). The use of stable and easily handled N,N-diisopropylphosphoramidites such as 2.53 (117, 118) is important for the automation of the process (119). The base-labile
|
|
|
O |
HO |
|
O |
NB(P1) |
|
|
|
|
|
|
DMTO |
|
|
|||
|
CPG |
L |
|
O |
+ |
|
|
|
|
|
|
|
O |
|
|
|
|||
|
|
O |
|
3' |
O |
|
|
|
|
|
|
|
P O |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NB(P1) |
iPr2N |
|
|
|
|
|
2.52 |
|
2.53 |
|
|
|
||
|
|
|
|
CN |
|
|
|||
|
|
|
|
|
DMTtO |
|
|
O |
R |
|
|
|
|
|
|
|
|
||
|
|
|
|
O |
O |
|
O |
O |
|
|
|
|
|
P |
O |
|
|
||
|
|
L O |
|
+ CPG |
L |
|
|
||
a, b |
|
O |
|
O |
|
||||
|
|
CPG |
3' |
O |
NB(P1) |
O |
3' |
O |
|
|
|
|
|
||||||
|
|
|
|
|
CN |
|
|
|
|
|
|
|
|
NB(P1) |
|
2.54 |
|
NB(P1) |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
capped unreactive |
|
|
|
|
|
|
c |
|
|
starting material |
|
|
|
|
|
|
DMTtO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
O |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
P |
O |
|
|
|
|
|
L |
|
|
|
|
|
|
|
|
|
O |
|
O |
|
|
|
|
|
|
|
CPG |
O |
|
|
|
|
|
|
|
|
3' |
|
NB(P1) |
|
|
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
CN |
|
|
|
|
|
|
|
|
NB(P1) |
|
|
|
|
|
|
|
|
|
d |
|
|
|
|
|
|
|
|
|
DMTtO |
|
|
|
|
|
|
|
|
|
O |
O |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
P |
O |
|
|
|
|
|
|
|
|
3' |
|
|
|
|
|
HO |
O |
OH |
|
||
|
NB(P1) |
|
|
|
NB(P1)
L = LINKER
a: 1H-tetrazole; b: capping, (RCO)2O or RCOCl; c: I2, pyridine, H2O; d: aq. NH4OH, heat.
Figure 2.17 Detailed phosphoramidite-based oligonucleotide SPS protocol.
66 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
β-cyanoethoxy protecting group is used for the phosphate function of 2.53 (120) and allows the simultaneous deprotection of this function and cleavage of the ON from the supports in the final step; other protected phosphoramidites were recently reported (121). Capping, that is, quenching of unreacted termini with either an acid anhydride or an acid chloride after each coupling cycle (step b, 2.54, Fig. 2.17), terminates all the sequences that would produce shortened ONs (122). Even if the yield of the coupling is typically >98%, the introduction of a capping step significantly improves the quality of the SPS and is especially important for long ON sequences. The oxidation with aqueous iodine is performed after rather than before each capping step, reducing further the amount of impurities (112).
The monitoring of a reaction and the methods used in determination of the structures of the products are influenced by the nature of the nonswelling CPG support, which does not allow the acquisition of NMR spectra in the solid phase. Other on-bead or off-bead methods presented in Sections 1.3 and 1.4 are more suited to the characterization and monitoring of the synthesis. When DMTt is used as the 5′-protecting group, the reaction can be monitored by quantitating the release of the bright orange trityl carbocation in solution by spectrophotometry at 498 nm (104) in a similar manner to the Fmoc method used in peptide synthesis.
2.2.2 Solid-Phase Synthesis of Oligonucleotide-Related Oligomers
There are several drawbacks to ONs as potential drugs that seriously limit their potential therapeutic applications; however, their potential to be selective and potent agents acting on specific targets is very high. Major hurdles for the exploitation of ONs are their susceptibility to endoand exonucleases, their poor solubility, and their poor pharmacokinetic properties.
Much effort has been expended toward the stabilization of ONs to render them more bioavailable while maintaining levels of selectivity and potency in binding to complementary nucleotide strands. A recent review (123) covers the preparation of analogues containing either modified nucleobases such as 2.55–2.57 or modified sugars 2.58– 2.60 (Fig. 2.18). Newer approaches have included replacement of the sugars with a 4-hydroxy-N-acetylprolinols 2.61 (124) and 2′-O,3′-C-linked [3.3.0] or [3.2.0] bicycloarabino sugars such as 2.62 (125) and 2.63 (126). All of these analogues are accessible by SP and possess interesting biological and physicochemical properties. Recently several phosphoramidite building blocks suitable to introduce nucleobaseor sugar-modified ON building blocks were reported; among them 2′-C-β-methyl- cytidine (127), 4,8-dihydro-4-hydroxy-8-oxo-2′-deoxyguanosine (128), several 7- substituted 7,8-dideazaadenines (129) and (5′S,6S)-5′,6-cyclo-5,6-dihydro-5, 6-dihydrothymidine (130).
Postsynthetic transformation of the nucleobases has also been reported using modified building blocks containing o-nitrobenzyl-protected acids or amines that after SP assembly and photolysis, could be selectively functionalized (131) or applying Pd-catalyzed cross-coupling reactions in carefully controlled reaction conditions on support-bound, preassembled ONs (132). Postsynthetic glycosidation of 5′-hydroxyls was also reported to give sugar-functionalized oligonucleotides (133).
|
2.2 |
OLIGONUCLEOTIDES 67 |
O |
NB |
|
HO |
|
|
|
|
|
HO |
|
|
O N X |
H |
H |
O N O |
O N N |
|
N |
NH |
N |
R1 |
|
X |
2.55 |
2.56 |
X=O, NH |
|
|
|
X=OH, NH2 |
|
2.57 |
R1=Me, Br, 2-pyridyl, 2-thiophenyl, |
|
|
2-thiazolyl, 2-imidazolyl, 1-propinyl. |
|
|
|
|
|
|
|
|
X |
|
O |
NB |
|
O |
NB |
NB |
|
HO |
|
||||
HO |
|
|
|
|
HO |
|
HO |
OX |
HO |
X |
|
HO |
|
|
2.58 |
|
|
2.59 |
|
2.60 |
X=alkyl, subst. alkyl, aryl |
X=alkyl, subst. alkyl, |
X=alkyl, OH, O-alkyl |
||||
|
|
|
aryl, alkenyl |
|
|
|
|
NB |
|
|
|
|
|
|
|
O |
O |
|
|
O NB |
|
|
|
NB |
|
||
|
N |
|
HO |
|
||
HO |
|
|
|
HO |
||
|
|
HO |
|
|
HO |
|
|
|
|
|
|
||
|
|
|
|
O |
|
O |
|
|
|
|
|
|
|
|
|
OR |
|
|
|
|
|
2.61 |
2.62 |
|
2.63 |
||
Figure 2.18 Modified nucleobases (2.55–2.57) and sugars (2.58–2.63) for the SPS of oligonucleotide analogues.
Modifications of the phosphodiester backbone have also been reviewed (123) and include phosphorus-containing replacements such as 2.64–2.66 (Fig. 2.19). Among these examples, the phosphorothioates 2.65 (134) and the phosphoramidates 2.66 (135) are particularly interesting as biologically active oligonucleotide analogues; several groups are working to improve the automated SPS of these and similar backbone-modified ONs (136, 137). The synthesis of a 15-mer phosphorothioate conjugate is reported in Section 2.2.3 as an example.
Cyclic ONs of small–medium size using the phosporamidite protocol to grow the ON chain and simple cyclization protocols to join the precursor ends were reported
68 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
|
|
HO |
|
HO |
|
HO |
|
|
X |
|
O |
|
|
O H |
|
|
O |
O |
O |
O |
|
O N |
|
|
P |
O |
P |
O |
|
P |
O |
|
|
S |
|
O |
|||
HO |
Y |
HO |
|
HO |
|
||
|
O |
NB |
O |
NB |
|
O |
NB |
|
|
|
|
|
|||
|
NB |
|
NB |
|
|
NB |
|
|
2.64 |
|
2.65 |
|
2.66 |
|
|
X=O, S Y=Me, NHR, OR, S |
phosphorothioates |
|
phosphoramidates |
|
|||
|
H |
|
|
|
|
NB |
|
|
N |
|
|
|
Me |
|
|
|
|
NB |
|
|
|
|
|
|
|
|
|
Si |
|
|
|
|
|
N |
|
DMTtO |
|
|
|
|
|
|
|
|
|
||
|
|
HO |
|
|
O |
|
|
|
|
N |
|
|
P |
O |
|
|
|
O |
NB |
|
|
||
|
|
iPr2N |
|
|
|||
|
|
N |
|
|
|
||
|
|
O |
|
|
|
CN |
|
|
|
2.67 |
|
|
2.68 |
|
|
O
peptide nucleic acids PNA
Figure 2.19 Phospodiester backbone replacements (2.66–2.68) commonly used for the SPS of oligonucleotide analogues.
(138, 139). The so-called peptide nucleic acids (PNAs) 2.67 (140–142) are examples of the replacement of the entire sugar–phosphate backbone with different functionalities that retain biological activity. The chemistry is similar to SP peptide synthesis employing Boc or Fmoc protocols (see Section 2.1). Peptide–ON hybrids, where either one (143) or two (144) of the ON termini are attached to peptide chains, have been reported. Acyclic silicon phosphoramidites 2.68 (145) have also been used for SPS of silicon-containing ON analogues.
A recent review (146) examines the features of several synthetic biopolymers mimicking the structures of both peptides and nucleotides and critically analyzes the future opportunities for each unnatural oligomeric family of compounds.
2.2.3 An Example: Synthesis of a Bioreversible ODN PS Conjugate
The synthesis of the target ON conjugate 5′-XTCTCACTACCTCTT (X = 2.72, Fig. 2.20) was performed using the SP phosphoramidite protocol with PNT (N-pent-4- enoyl)-protected phosphoramidites 2.69 and 2.70, the unprotected phosphoramidite 2.71, and the conjugated phosphoramidite 2.72 (147). Their structure and synthesis (76) from natural nucleosides is reported in Fig. 2.20. The protection of the base was
2.2 OLIGONUCLEOTIDES 69
|
|
|
|
|
|
|
O |
|
N |
|
NH2 |
|
|
|
|
NH2 |
|
|
N |
|
|
||
O |
|
|
N |
|
HO |
|
|
|
|||
|
|
N |
|
|
N |
N |
|
||||
HO |
|
|
|
|
|
HO |
|
|
|||
|
|
|
|
|
|
|
|
|
|
||
|
|
|
O |
|
|
|
|
|
|
|
|
HO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a,b,c |
|
|
|
|
|
|
a,b,c |
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
N |
|
H |
O |
|
|
|
|
N |
O |
|
O |
N |
|
N |
|
|
|
O |
|
|
|
|
|||||
|
|
N |
|
DMTtO |
|
|
|
|
|||
|
|
N |
|
|
|
|
|
|
|||
DMTtO |
|
|
|
|
|
N |
|
|
|||
|
|
|
|
|
|
N |
|
|
|||
|
|
|
|
|
|
O |
|
|
|||
|
|
|
|
O |
|
|
|
|
|
||
O |
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
P O |
|
|
|
|
|
P |
|
O |
|
|
|
|
|
|
|
||
|
|
|
iPr |
N |
|
|
|
|
|||
iPr2N |
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
CN |
|
|
|
|
|
|
|
CN |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
||
|
2.69 |
|
|
|
|
2.70 |
|
|
|
||
|
|
|
|
O |
|
|
O |
|
|
O |
|
O |
|
|
|
|
|
N |
|
|
|
||
|
|
N |
|
|
|
NH |
|
||||
|
|
NH |
|
HO |
|
||||||
HO |
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
O |
|
|
|
|
HO |
|
|
O |
|
|
|
HO |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
b,c |
|
|
|
|
b,d |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
O |
|
|
O |
|
|
O |
|
|
|
N |
NH |
|
|
N |
NH |
|
|
|||
DMTtO |
|
|
|
DMTtO |
|
|
|
||||
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
||
O |
|
|
O |
|
|
|
O |
O |
|
|
Me |
|
|
|
|
|
|
|
|
O |
|||
P O |
|
|
|
|
P O |
|
|
|
|||
|
|
|
|
|
|
|
|
||||
iPr2N |
|
|
|
|
|
iPr2N |
|
|
O |
|
|
|
|
|
CN |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
Me |
||
|
|
|
|
|
|
|
|
|
|
|
|
2.71 |
|
|
|
|
2.72 |
|
|
||||
|
|
|
|
|
|
|
|
|
|
||
a:TMSCl, pyridine, pent-4-enoyl anhydride, 3 hrs, rt;
b:DMTt-Cl, pyridine, 1.5 hrs, rt; c: amidite, TEA, DCM, 3hrs, rt;
d:di-iPr phosphorus dichloride, 4-(2,6-dimethylbenzoyloxy)-benzyl alcohol, TEA, DCM, 40', 0°C to rt.
Figure 2.20 Structure and synthesis of nucleotidic building blocks 2.69–2.72.
carried out through the corresponding anhydride (step a), followed by protection of the 5′-OH with DMTt-Cl (step b). The protected nucleoside was phosphitylated with a suitable chlorophosphoramidite (step c) to give the building blocks 2.69–2.70. The thymidine-based building block 2.71 did not require nucleobase protection and was prepared by submitting thymidine to steps b and c. The conjugated phosphoramidite

70 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
2.72 was prepared from 5′-protected thymidine (step b) in a one-pot reaction with diisopropyl phosphorus dichloride and 4-(2,6-dimethyl)benzoyloxybenzyl alcohol (step d).
The SP assembly of the ON 15-mer was performed on a 1-µmol scale using an automated synthesizer. The succinate was coupled to the LCAA-CPG support (500 Å) to give 2.73 (Fig. 2.21), which was submitted to reaction cycles for each elongation. The first elongation cycle is shown in detail in Fig. 2.21 and begins with detritylation of the resin-bound intermediate (step a) followed by a washing step (step b). Coupling
|
|
|
|
|
|
|
O |
HO |
|
|
|
|
|
|
|
|
|
|
CPG |
|
L |
|
|
|
3' |
|
|
|
|
O |
|
|
|
O |
|
|
|
|
|
|
|
O |
|
|
|
|
N |
|
N |
|
|||
|
|
|
|
O |
|
|
|
O |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
+ |
DMTt |
O |
|
|
|
|
|
|||
|
|
|
|
2.73 |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
O |
|
|
|||||
|
|
|
|
|
|
|
|
N |
|
2.71 O |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
O |
|
|
|
P |
N |
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
DMTt |
O |
|
CN |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
S |
O |
|
|
|
|
|
|
|
dC dAdT dC |
||
|
|
|
|
3' |
P |
|
|
L |
|
3' |
dT |
||||||
|
|
|
L |
|
|
O |
O |
a-h |
|
|
dT dC |
|
dC |
dA |
|||
b-h |
|
|
O |
|
|
|
CPG |
|
dT |
|
|
dT |
|||||
CPG |
|
|
O |
|
|
|
|
|
|
|
dT dC |
||||||
|
|
|
|
|
T |
13 |
|
|
|
5' |
|
O |
|
dC |
|||
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
||||
|
|
|
|
|
|
|
|
CN |
cycles |
|
|
|
P |
|
|
||
|
|
|
2.74 |
|
T |
|
|
|
DMTt |
O |
dT O |
O |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3' |
|
|
dC dAdT dC |
|
|
Me |
O |
|
|
|
|
||||
|
HO |
|
dT |
a, i |
|
|
O |
|
|
|
|||||||
|
|
dT dC |
dC |
|
|
dA |
|
|
|
|
|
|
|||||
|
|
dT |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
dT |
|
dT dC |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
dC |
|
|
|
|
|
Me |
|
|
|
|
|
|
|
|
5' |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
2.75 |
|
dTS P |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
HO |
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Me O
O
Me
L = LINKER
Automated reaction cycle (1mm-scale, continuous-flow):
a:3% trichloroacetic acid/DCM, 50 sec, 0.75 mL delivered;
b:wash with MeCN, 20 sec, 0.82 mL delivered;
c:0.08M 2.69-2.72/0.5M tetrazole/MeCN, 45 sec, 0.45 mL delivered;
d:wash with MeCN, 56 sec, 0.50 mL delivered;
e: 5% 3H-benzodithiole-3-one-1,1-dioxide/MeCN, 8 sec, 0.30 mL delivered;
f:wash with MeCN, 98 sec, 1.50 mL delivered;
g:acetic anhydride/2,6-lutidine/THF 1/1/18, 16% NMeImidazole/THF,
15 sec, 0.30 mL delivered;
h: wash with MeCN, 98 sec, 1.50 mL delivered.
i: K2CO3 0.05M, MeOH, 8 hrs, rt.
Figure 2.21 SPS of the ON phosphorothioate conjugate 5′-XTCTCACTACCTCTT. 2.75.
2.3 OLIGOSACCHARIDES 71
with a suitable phosphoramidite catalyzed by 1H-tetrazole (step c), another washing (step d), and then oxidative sulfurization (step e) with 3H-benzodithiole-3-one-1,1-di- oxide (148), again followed by washing (step f), gave the elaborated oligonucleotide 2.74. The unreacted sites on the support were capped (step g) and the resin was washed (step h) prior to iteration for 13 cycles. The conditions described were readily applied to automated continuous-flow SPS allowing for an extremely rapid and reliable preparation of the resin-bound target molecule (147). The simultaneous deprotection of the PNT group and the β-cyanoethyl group and cleavage from the solid support were finally carried out under mild conditions by treatment with methanolic potassium carbonate to give the target 2.75 (step i, Fig. 2.21).
The examples of SPS presented here and in Section 2.1.3 take advantage of multistep procedures that are routinely performed using an automated SP–peptide or SP–oligonucleotide synthesizer with reduced expenditure on optimization of conditions. Similar results with limited effort can be obtained for any oligomeric SPS (e.g., peptides, peptoids, oligonucleotides, oligoureas), providing that the repeating units form a backbone that is stable and robust during all of the synthetic steps. If the target molecule is not made up of repeating units but must be prepared by multistep SPS using diverse chemical reactions, the complexity of the SPS increases and the optimization of the chemistry on the SP becomes both crucial and time consuming, as we will see in the following chapter.
2.3 OLIGOSACCHARIDES
2.3.1 Solid-Phase Synthesis of Oligosaccharides
The SPS of oligosaccharides (OSs), like that of peptides and oligonucleotides, started in the early 1970s (149, 150) with the demonstration that the coupling between monosaccharide units was feasible on a solid support using different protocols (151– 153). The higher complexity of the sugar scaffold, though, prevented the development of a reliable SP procedure for the assembly of carbohydrate building blocks in a regioand stereospecific manner. In general, the multiple reactive sites and the stereochemical complexity have often hampered OS synthesis and, as of this writing, have prevented the growth of an automated technology for the SPS of OSs.
These limitations notwithstanding, the past 20 years have witnessed major achievements in SP OS synthesis, which now allows the reliable assembly of small oligomers (up to four to five saccharide units) in most of the cases with good yields and purity. A few glycosylation technologies for oligomer growth on SP that use different functionalities are available and among them the chemist may choose according to the needs of the specific synthesis; for example, the glycal (154), the sulfoxide (155), the trichloroacetylimidate (156), and the enzyme-based technology (Fig. 2.22) (157) have all been successfully employed. We will briefly describe the salient points of each method and show their usefulness through a critical review of their features with regard to the formation of α- or β-anomers, coupling with hydroxyls having different

72 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
|
O |
|
O |
S O |
|
|
||
GLYCAL |
|
|
|
SULFOXIDE |
|
O |
|
|
Cl |
ENZYMATIC |
|
O |
||
GLYCOSYLATION: |
||
Cl |
|
|
Cl |
PURIFIED ENZYME + |
|
NH |
||
NATURAL SUBSTRATE |
||
|
||
TRICHLOROACETYLIMIDATE |
|
Figure 2.22 Common SP glycosylation reagents and methods.
reactivities and protecting groups that are compatible with their use (Fig. 2.22, bottom); a list of major issues to consider for OS SPS is reported in Fig. 2.23.
The glycal technology was first reported on SP by Danishefsky et al. (154) following a typical procedure reported in Fig. 2.24. The supported glycal 2.76, where C6–OH is used to link the glycal to the resin, was prepared in three steps from PS resin and was then epoxidized with DMDO (dimethyl dioxirane) to give the resin-bound glycosyl donor 2.77. Treatment of 2.77 with a solution of a suitably protected glycosyl acceptor A in the presence of anhydrous zinc chloride produced the β-(1→6) galactosyl disaccharide 2.78, which was further elaborated to the tetrasaccharide 2.79 using the same chemistry with different glycosyl acceptors (A and B, Fig. 2.24). Each free C2–OH that originates from any glycosylation could be kept unprotected and eventually be used as a further reaction site (vide infra). The galactosyl anhydrosugar proved to be a suitable donor even for hindered glycosyl acceptors, but the glucosyl analogue proved to be unsuitable for all of the desired transformations due to side reactions, especially when coupled to less reactive acceptors. The elaboration of the resin-bound glycal 2.80 to a protected thioethyl donor 2.83 (158) via the anhydroglucose 2.81
OH
O
HO
HO
OH OH
-anomeric carbon / stereochemistry?
-OH reactivities / regiochemistry? - orthogonal protecting groups?
-support/linkers/cleavage compatible with OS structure?
-common, automation-friendly protocols?
Figure 2.23 Major issues in SP OS synthesis.