

Solid-Phase Synthesis and Combinatorial Technologies
.pdf2.3 OLIGOSACCHARIDES 73
|
|
Ph |
|
|
|
|
Ph |
|
|
|
a,b |
|
Si |
|
c |
|
|
O |
O |
Si |
|
|
Cl |
|
|
|
|
|||||
|
|
|
|
|
|
O |
O |
Ph |
|
|
|
|
Ph |
|
|
|
O |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2.76 |
|
|
Ph |
|
|
|
|
|
|
O O SiPh2PS |
||
d |
O |
Si |
|
|
e |
|
O |
|
O |
|
O |
|
|
|
O |
|
O |
||||
O |
O Ph |
|
|
|
|
|
OH |
|||
|
|
|
|
|
|
|
O |
|||
|
O |
|
|
|
|
|
|
|
O |
|
|
|
O |
2.77 |
|
|
|
|
2.78 |
O |
|
|
|
|
|
|
|
|
|
O |
||
|
|
|
|
|
|
|
|
|
|
|
O O SiPh2PS |
|
|
|
O OH |
|
|
|
|||
|
|
O |
|
|
O |
|
|
|
||
O |
O |
|
|
|
O |
|
OH |
O |
|
|
O |
O |
|
|
|
|
|
O |
|
|
|
|
OH O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
O |
|
|
d,e,d,f |
O |
O |
O |
|
|
g |
O |
O |
|
|
O |
|
|
|
|
|
OH |
|
|||
|
|
|
|
|
|
|
|
|||
|
|
OH O |
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
O |
O |
|||
|
|
O |
|
|
O |
|
O |
|
O |
O |
|
|
|
O |
|
|
|
|
|
OH |
|
2.79OH
|
O |
BnO |
O |
BnO |
|
||
BnO |
|
||
BnO |
|
|
|
O |
OH |
OH |
|
|
|||
O |
O |
|
|
O |
O |
||
BnO |
|||
|
BnO |
|
|
A |
|
B |
|
|
|
a: nBuLi, TMEDA, cyclohexane; b: Ph2SiCl2,
benzene; c: A, DIPEA, DCM; d: DMDO, DCM; e: A, ZnCl2, THF; f: B, ZnCl2, THF; g: TBAF,
AcOH, THF.
Figure 2.24 SP glycal protocol: SP synthesis of 2.79.
allowed the formation of the α-glucosidic linkage 2.84 (Fig. 2.25). The functionalization of the less reactive C2–OH by using the thioethyl donor in solution after removal of the pivaloyl protecting group on SP was also used to give the branched trisaccharide 2.86 (Fig. 2.25). Removal of the protecting groups used during the synthesis and cleavage from the support produced the free C6–OH saccharide (vide infra). The glycal technology thus allows the construction of diversified oligosaccharides with various possible connections (159), including N-linked glycopeptides (160) and even branched oligomeric chains (161, 162). Unfortunately the α-stereochemistry at the anomeric center cannot be obtained as major glycosylation products with these protocols (163).

74 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
|
iPr |
|
|
|
|
|
|
|
|
|
|
|
Si |
a |
|
Pri |
|
b |
|
Pri |
|
|
|
|
|
O |
|
|
O |
|
|
|
|||
|
Cl |
|
|
Si |
|
|
Si |
|
|
||
|
iPr |
|
|
|
|
|
|
|
|
||
|
|
|
BnO |
O |
iPr |
2.80 |
BnO |
O |
iPr |
2.81 |
|
|
|
|
BnO |
|
|
BnO |
|
O |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
iPr |
|
|
|
|
iPr |
|
|
iPr |
|
|
c |
Si |
|
|
|
|
Si |
|
|
|
|
|
O iPr |
|
|
d |
|
O iPr |
e |
Si |
|
|
||
|
|
|
|
|
OBn |
||||||
|
O |
|
|
BnO |
|
O |
|
O |
iPr |
|
|
|
BnO |
|
SEt |
|
SEt |
|
|
O |
|
|
|
|
BnO |
|
BnO |
BnO |
|
O |
O |
||||
|
|
|
OPiv |
|
|
||||||
|
OH |
|
|
|
BnO |
|
|
|
|||
|
2.82 |
|
|
|
2.83 |
|
|
OPiv O |
|
||
|
|
|
|
|
|
|
|
2.84 |
Bn |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
iPr |
|
|
|
|
g |
iPr |
|
|
|
|
|
f |
|
|
|
|
OSi |
|
|
|
|
|
|
OSi |
|
|
OBn |
|
|
|
|
OBn |
|
|
|
iPr |
|
|
|
|
iPr |
|
|
|
|
|
|
O |
|
|
|
|
BnO |
O |
|
|
O |
|
|
BnO |
|
O |
O |
|
|
O |
|
|
||
|
|
|
BnO |
O |
|
|
|
||||
|
BnO |
|
O |
|
|
BnO |
O |
|
|
||
|
OH |
Bn |
|
|
O |
Bn |
|
|
|
||
|
2.85 |
|
|
|
BnO |
2.86 |
|
||||
|
|
|
|
|
|
||||||
|
|
|
|
|
|
BnO |
OPiv |
59% from 2.80 |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
a:A, DIPEA, DMAP, DCM, rt, 72 hrs; b: DMDO, DCM, 0°C, 2.5 hrs; c: EtSH, Tf2O, -78°C to rt, DCM;
d:PivCl, DMAP, DCM, rt, 4 hrs; e: B, MeOTf, DTBP, 4A MS, DCM, 0°C to rt, 8 hrs;
f:DIBAL, DCM, -78°C, 5 hrs; g: C, MeOTf, DTBP, 4A MS, DCM, 0°C to rt, 8 hrs.
A |
OH |
B |
OCH2Ph |
PhCH2O |
C |
|
|
|
|||||
|
O |
|
O |
O |
||
PhCH2O |
HO |
PhCH2O |
||||
|
SEt |
|||||
PhCH2O |
|
PhCH O |
|
PhCH O |
||
|
|
2 |
|
2 |
OPiv |
|
|
|
|
|
|
Figure 2.25 SP glycal protocol: thioethyl donors for the synthesis of 2.86.
The sulfoxide technology was first reported by Yan et al. (155); its main features are shown schematically in Fig. 2.26. The resin-bound glycosyl acceptor 2.87, easily prepared from Merrifield PS resin and the p-hydroxythiophenyl glycoside (164), was coupled with two different donors A and B in the presence of triflic anhydride and a hindered base. The two couplings produced respectively the α- and the β-linked disaccharides 2.88 and 2.89 in good yield. The presence of a C2-neighboring group able to participate in the glycosylation, such as the pivaloyl in B, assured the β-stereochemistry while the absence of such a group combined with low reaction temperature (–60 °C) invariably gave α-selectivity with secondary alcohols. Removal

|
|
|
|
|
|
2.3 |
OLIGOSACCHARIDES 75 |
|
|
|
|
|
|
O |
|
|
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
O |
O |
|
2.87 |
|
|
|
|
|
O HO |
|
S |
|
|
|
|
|
a |
|
N3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
Ph |
|
|
|
|
b |
|
|
|
|
O |
O |
|
|
|
|
|
|
|
O O |
N3 |
S |
|
|
|
|
|
|
|
2.88 |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OBn |
|
|
|
|
|
|
|
O |
O |
|
|
|
|
|
|
|
Bn |
|
|
|
|
|
|
O |
|
Bn |
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
a: A, Tf2O, DTMP, DCM, -60° to -30°C, 1 hr; |
|
OPiv |
O |
|
O |
|||
Piv |
O O O |
|
S 2.89 |
|||||
b: B, Tf O, DTMP, DCM, -60° to -30°C, 1 hr. |
O |
|
||||||
2 |
|
|
|
|
|
|
|
N3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PivO |
OPiv |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O Ph |
|
Piv |
|
|
|
|
|
|
S |
|
|
|
|
|
|
|
|
|
|
O OPiv |
|
O |
|
|
|
|
O |
|
|
O |
|
|
|
|
|
|
|
|
S |
||
|
|
O |
OBn |
|
PivO |
|
|
|
|
|
|
|
B OPiv |
Ph |
|||
|
|
O Bn |
|
|
|
|||
|
Bn |
A |
|
|
|
|||
|
|
|
|
|
|
|
||
Figure 2.26 SP sulfoxide protocol: synthesis of 2.88 and 2.89.
of the protecting groups and cleavage from the support produced the free reducing saccharide as an anomeric mixture (vide infra). Other SP sulfoxide-based syntheses have been reported (165), including an SP library preparation (166), and the method appears to be both flexible (e.g., both α-, β-, and diversely structured oligosaccharides can be prepared) and reliable.
The trichloroacetimidate route was first reported on soluble supports by Douglas et al. (167) and then on PS supports by Rademann et al. (156, 168) with the general scheme shown in Fig. 2.27. The polymer-bound protected acceptor 2.90 was prepared from the corresponding trichloroacetimidate A and an SH-functionalized PS resin. After deacetylation it was coupled with A in the presence of trimethylsilyl trifluoromethanesulfonate as catalyst to give the 1,2-trans α-dimannoside 2.92, which was extended to the pentamannoside 2.93 by repeating three glycosylation/deacetylation cycles (Fig. 2.27). The free reducing saccharide or its methyl derivative (vide infra) was produced after deprotection and cleavage. Several other reports of trichlo-

76 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
|
a |
BnO |
O Ac |
|
O |
||
O |
SH |
BnO |
|
BnO |
|
||
|
|
|
|
|
|
OH |
2.90 S |
|
|
|
|
|
|
BnO |
O |
|
|
||
|
b |
|
|
O |
|
|
||
|
BnO |
|
|
|
|
|||
|
|
|
|
|
|
|
||
|
|
|
BnO |
|
|
|
|
|
|
|
|
|
2.91 |
S |
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
BnO |
O Ac |
|
|
|
||
|
|
O |
|
|
|
|
||
|
BnO |
|
|
|
|
|
||
a |
|
|
|
|
|
|
||
BnO |
|
|
|
|
|
|||
|
|
|
BnO |
O |
|
|
|
|
|
|
|
O |
|
|
|
||
|
|
BnO |
|
|
|
|||
|
|
|
|
|
|
|
||
|
|
|
BnO |
|
2.92 |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
S |
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
BnO |
O Ac |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
BnO |
|
|
|
|
|
|
|
|
|
|
|
O Ac |
|
|
|
|
|
BnO |
|
|
BnO |
|
|
|
|
|
BnO |
O |
O |
|
||
|
|
|
BnO |
|
||||
|
|
|
O |
|
|
|||
|
|
|
BnO |
|
BnO |
|
|
|
|
|
|
BnO |
|
|
O |
NH2 |
|
|
b,a,b,a,b,a |
|
|
BnO |
O |
A |
||
|
|
|
Cl |
|
||||
|
|
|
BnO |
O |
|
|
||
|
|
|
|
|
Cl |
Cl |
||
|
|
|
|
BnO |
|
|
|
|
|
|
|
|
BnO |
O |
|
|
|
|
|
|
|
BnO |
O |
|
|
|
|
|
|
|
BnO |
|
|
|
|
|
|
|
|
|
BnO |
O |
|
|
|
|
|
|
BnO |
O |
|
|
|
|
|
|
|
2.93 |
|
|
||
|
|
|
|
|
BnO |
|
|
|
S
O
a: A, TMSOTf, DCM, rt, 1 hr; b: 0.5M NaOMe, DCM/MeOH 10/1, rt, 2 hrs.
Figure 2.27 SP trichloroacetimidate protocol: synthesis of 2.93.
roacetimidate SP glycosylation followed (169, 170) where both α- and β-stereochem- istries were introduced in the same complex oligosaccharide structure, thus proving this method to be versatile enough for various SP synthetic applications.
Enzymatic glycosylation on SP was first reported by Schuster et al. (157) and since then has been used to perform multiple glycosylations. Two examples are reported in Fig. 2.28. The synthesis of oligosaccharides 2.94 and 2.95 related to the sialyl Lewis X antigen was carried out via glycosylations mediated by β-1,4-galactosyltransferase and α-2,3-sialyltransferase (171) and by β-1,4-galactosyltransferase, α-2,3-sialyl- transferase, and fucosyltransferase (172), respectively. The use of unprotected saccha-

|
|
|
|
|
|
|
2.3 |
OLIGOSACCHARIDES 77 |
|
|
|
|
OH |
OHOH |
|
|
O |
|
|
|
|
|
O |
|
O |
|
H |
|
|
|
|
HO |
|
|
|
|
|||
|
|
O |
O |
|
|
N |
|
||
|
|
HO |
OH |
|
|
O |
CPG |
||
|
|
NHAc |
|
|
|
||||
|
|
|
|
|
|
|
O |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a |
|
|
|
|
|
OH |
OH |
OHOH |
|
|
|
|
|
|
|
O |
|
|
O |
|
|||
|
HO |
|
O |
O |
|
|
H |
||
|
|
O |
O |
|
|
||||
|
HO |
|
O |
|
|
|
N |
||
|
|
OH |
HO |
NHAc |
OH |
|
|
O |
CPG |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
b |
|
|
|
|
OH |
HO |
O OH OH |
|
OH |
OHOH |
|
O |
|
HO |
OH |
|
O |
|
O |
|
O |
H |
|
|
|
|
O |
|
|
||||
AcNH |
O |
O |
|
O |
OH |
O |
N |
||
|
HO |
|
OH HO |
NHAc |
(5) O |
CPG |
|||
O
2.94
a: UDP-Galactose, β-(1-4)galactosyltransferase; b: CMP-Neu5Ac, α-(2-3)sialyltransferase.
|
OH |
HO |
O OH OH |
|
OH |
|
|
|
HO |
OH |
|
O |
|
O |
|
|
|
|
O |
O |
O |
|
|
|
||
AcNH |
O |
|
|
S |
||||
HO |
|
|
||||||
|
OH |
|
|
|||||
|
HO |
|
|
|
NHAc |
N |
|
S |
|
|
|
|
|
|
O |
SEPH |
|
|
|
|
|
|
|
H |
||
|
|
|
|
|
c |
|
|
|
|
OH |
HO |
O OH OH |
|
OH |
|
|
|
HO |
OH |
|
O |
|
O |
|
|
|
|
O |
O |
O |
|
|
|
||
AcNH |
O |
|
|
S |
||||
O |
|
|
||||||
|
OH |
|
|
|||||
|
HO |
|
|
|
NHAc |
N |
|
S |
|
|
|
O |
|
|
O |
SEPH |
|
|
|
|
OH |
2.95 |
H |
|||
|
|
|
|
|
|
|
||
HO
OH
c: GDP-fucose, α-(1-3/4)fucosyltransferase
Figure 2.28 SP enzymatic glycosylation protocol: synthesis of 2.94 and 2.95.
ride substrates, the specificity of the enzymes, and the availability of complex structures, which are currently too challenging for organic chemistry, are the main advantages of this method. However, the limited availability of the necessary enzymes, the constraints of using natural substrates for each purified enzyme, and the slow kinetics of SP enzymatic glycosylations are the drawbacks that confine this approach to specific applications where even kilograms of complex saccharide derivatives have been obtained (173).
A variety of solid supports have been used for OS SPS, including PS resins (155, 161, 170, 174), PS crowns (175), soluble PEG supports (167, 176, 177), CPG (169,

78 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
171), and sepharose (172). Each OS SP assembly method has preferentially adopted a specific support, most probably because of the expertise of the leading research group in the field. A variety of linkers have also been used and some of them are shown in Fig. 2.29 (2.96–2.100) and 2.30 (2.101, 2.102 and 2.105). The Si-based linker 2.96 (156, 158–162) was prepared in three steps from PS resin and the glycal derivative A, then finally cleaved by fluoridolysis to give the pure α or β anomeric saccharide. Another similar, commercially available Si linker 2.97 used in SP (see Chapter 1, Fig. 1.8, 1.10) was used in OS SPS (178) and cleaved with AcOH–THF–H2O cocktails at 65 °C recovering pure disaccharides in good yields. The Si-based linker 2.98 (179) was prepared from silyl alcohol B (180) and trichloroacetimidate C in five steps and finally cleaved with acetic anhydride–Lewis acid to give the corresponding α,β anomeric acetate. The S-based linker 2.99 (155, 164) was prepared from Merrifield resin and a suitable p-hydroxythiophenyl glycoside D (164) and finally cleaved by mercury salts to give the α,β anomeric free oligosaccharide. The photolinker 2.100 (181) was prepared in two steps from Merrifield PS resin and the β-phenolic ester E and finally cleaved by irradiation at rt to give the pure α or β anomeric phenoxy ester.
O |
|
R |
|
|
|
|
O |
Si |
a |
|
O OH |
|
|
|
|
|
||||
|
|
|
|
|
||
|
|
R |
|
|
A |
|
|
|
|
|
|
|
|
2.96 |
R = Ph; |
a: TBAF/AcOH 2/1, THF, 40°C, 18 hrs. |
||||
2.97 |
R = Et; |
a: AcOH/THF/H2O 6/6/1, 65°C, 10 hrs. |
|
|||
O |
|
|
O |
|
|
|
O |
|
|
|
|
b |
|
|
|
Si |
N |
|
||
|
|
|
|
|
||
|
|
|
|
H |
|
|
b: BF3.Et2O, Ac2O, toluene, rt, 23 hrs. |
|
|
HO |
|||
|
2.98 |
|
||||
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
O |
S |
|
2.99 |
c |
O |
OH |
|
|
|
|
|||
|
|
|
|
|
||
|
c: Hg(OCOCF3)2, DCM, H2O, rt, 5 hrs. |
|
||||
|
|
|
O2N |
|
|
|
|
|
|
O |
O |
|
d |
O |
|
|
|
|
||
O |
|
2.100 |
|
|
|
|
|
|
|
|
|
||
O
d: hν, THF, rt, 20 hrs.
|
O |
|
C |
O |
NH |
|
|
|
|
Cl |
|
|
Cl |
Cl |
O
 OAc
OAc
Si
B
OH
O
S D
OH
O
O
E O
Figure 2.29 Linkers used in SP OS synthesis: 2.96–2.100.

R1
S |
SPS |
R
2.101 R1 = OTMS, R2 = H
2.102 R1 = OTBDMS, R2 = OBn
BnO HN |
PMP OH |
|
OBn |
OH |
2.104 |
EtO
O
|
2.3 OLIGOSACCHARIDES 79 |
||
TBDS |
O HN |
PMP |
|
|
O |
|
|
BnO |
|
|
S |
|
|
|
|
|
O |
OBn |
|
TBDS |
|
|
|
|
2.103 |
|
|
|
|
e |
|
|
HO |
|
|
f |
|
O |
O e: TBAF, 12 hrs, rt; |
BnO |
|
||
|
f: DIBAL, DCM, |
||
|
|
||
|
|
|
-78°C. |
|
HN |
|
OBn |
|
PMP |
|
|
H
N
L
S S = sepharose
2.105
O
a,b,c
|
OH OH |
|
OH |
OH |
|
|
OH |
|
|
|
|
||
|
|
OH |
|
|
|
|
|
|
|
||||
|
O |
|
O |
|
O |
|
O |
O |
H |
H |
|
|
|
HO |
|
O |
|
O |
|
|
|
|
N |
N |
L |
|
|
OH |
HO |
NHAc |
|
OH |
HO |
OH |
|
S |
|||||
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
O |
O |
|
|
|
|
|
|
|
|
|
|
d or e |
|
|
|
|
|
|
|
OH OH |
|
OH |
|
|
OH |
|
|
OH |
|
|
|
|
|
|
|
|
OH |
|
|
|
|
||||
|
|
|
O |
|
O |
|
|
O |
O |
O |
OH |
|
|
|
HO |
O |
O |
|
|
||||||||
|
|
|
|
|
|
|
|||||||
|
OH |
HO |
|
NHAc |
OH |
HO |
OH |
|
|
||||
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|||||
a:SP glycosylation (SP disaccharide); b,c: enzymatic SP glycosylations (to SP tetrasaccharide);
d:Br2, H2O, 10', rt, then NaBO3, ph 8, 1 hr, rt; e: NaBO3, NH4OH, ovn., rt.
Figure 2.30 Linker used in SP OS synthesis: 2.101, 2.102 and 2.105.
An intriguing report dealing with various monosaccharides made on SP and cleaved using a cyclative cleavage (CC) strategy was based on the silyl enol ether linkers 2.101 and 2.102, easily obtained from thiol PS resin via acylation and O-alkylation; their SP elaboration to structures such as 2.103 was followed by cleavage using TBAF via TBDS (tert-butyl dimethyl silyl) deprotection and lactone formation to give, after
80 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
reduction, the sugar 2.104 (182). The squarate linker 2.105 (183), prepared on Sepharose beads, was both chemically and enzymatically glycosylated to give a tetrasaccharide that was cleaved in good yield and purity either with aqueous bromine or with ammonia/ammonium borate.
A variety of protecting groups have also been used for the saccharide hydroxyls, including esters (acetate, benzoate, pivaloate), ethers (benzyl), silyl groups (TBDPS (tert-butyl diphenyl silyl)), carbonates, and benzylidene acetals; protecting groups tailored for the specific needs of OS SP have also been reported (184). Their careful selection as orthogonal protections for different OHs and their general compatibility with the above-mentioned and other SP linkers have allowed the successful SP synthesis of many complex oligosaccharides.
Reaction monitoring and structure determination have often used the more reliable off-bead analytical characterization. Isomeric oligosaccharides have the same MW, thus hampering MS techniques, and their NMR spectra are quite complex. Nevertheless, examples of significant 1H MAS NMR spectra of resin-bound saccharides have been reported (185).
Several other glycosylation technologies have recently been reported, among them the methods by Nicolaou et al. (174, 181), Ito et al. (177), and Rodebaugh et al. (175) are worth mentioning and will probably become more widely used in the future. As a general comment, the extreme complexity of OS synthesis has required the development of a number of choices for the chemistry that are complementary rather than exclusive. The assessment of one, or more likely a few, universal SP routes for OS SP synthesis and their subsequent automation appears today to be an extremely ambitious, challenging, and eventually obtainable target, considering the increasing interest of key academic and industrial groups in SP oligosaccharide synthesis.
Several recent reviews (186–189) can provide the reader with more detail regarding all the previously examined aspects of oligosaccharide SPS. These reviews also cover the major aspects of oligosaccharide-related oligomers, which have either already been introduced, as for glycopeptides in Section 2.1.2 or for oligonucleotides in Section 2.2, or which will be described in other chapters, as for sugar-containing natural products described in Chapter 4 or for sugar scaffold analogues described as small organic molecules in Chapters 6–9.
2.3.2 Example: Synthesis of 3,4,6-Tri-O-benzyl-a-D-mannopyranosyl 3,4,6-tri-O-benzyl-a-D-mannopyranoside and 3,4-Di-O-acetyl-2-O-benzyl-a-L-fucopyranosyl 1-O-methyl-3,4,6-tri-O-benzyl-a-D-mannopyranoside
The synthesis of two 1,2 disaccharides 2.106 and 2.107, with α-mannose and α-fucose linkages, respectively, was reported by Rademann and Schmidt (168) using the trichloroacetimidate technology via the coupling of two different glycosyl donors, 2.109 and 2.110 (Fig. 2.31), to the PS-supported linker 2.108. The former was chosen because of the α-driving 2-OAc group (neighboring group participation in the glycosylation); the latter was chosen as a more challenging example for the generation of a 1,2-cis disaccharide. The synthesis of 2.108 is shown in Fig. 2.32. The two-step

|
2.3 |
OLIGOSACCHARIDES 81 |
||
|
|
Ac |
|
|
|
|
O Ac |
|
|
|
Ac |
O |
OBn |
|
BnO |
O |
O |
||
BnO |
O |
|
||
BnO |
|
|
|
|
|
O |
BnO |
O |
|
BnO |
BnO |
O |
||
O |
|
|||
BnO |
|
BnO |
|
|
BnO |
|
|
OMe |
|
2.106 |
OH |
2.107 |
||
|
||||
|
|
|
||
BnO |
Ac |
|
Ac |
Ac |
|
|
O |
|
O |
|
|
||
BnO |
O |
|
|
O |
|
|
|
|
SH |
OBn |
|
||
BnO |
|
|
|
|
||
|
|
|
O |
|
O |
|
2.109 |
O |
NH |
|
O |
NH |
|
Cl |
|
2.110 |
||||
|
|
|
||||
|
|
2.108 |
|
Cl |
|
|
|
Cl |
Cl |
|
|
|
|
|
|
|
Cl |
Cl |
||
|
|
|
|
|
||
Figure 2.31 Retrosynthetic SP scheme for compounds 2.106 and 2.107.
preparation of the S-protected linker A was followed by its coupling to Merrifield resin and final S-deprotection. The trichloroacetimidate glycosyl donors 2.109 and 2.110 were prepared from commercially available precursors using reported procedures (190, 191).
Cl |
+ |
|
COOMe |
a |
MMT |
COOMe |
MMT |
HS |
|
|
S |
||
|
|
|
|
|
|
|
b |
|
+ |
|
|
c |
|
MMT |
|
|
|
|
||
S |
|
OH |
|
Cl |
|
S MMT |
|
A |
|
|
|
O |
d
SH
O
2.108
a:pyridine, 14 hrs, rt; b: LiAlH4, 0°C, 2 hrs; c: NaH, 15-crown-5, THF, 60°C, 24 hrs;
d:washings with TFA/DCM 5/95, rt.
Figure 2.32 Synthesis of linker 2.108.
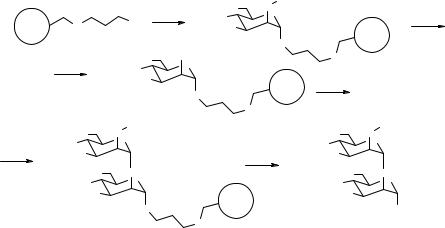
82 SOLID-PHASE SYNTHESIS: OLIGOMERIC MOLECULES
The synthesis of 2.106, which was partially described in Fig. 2.27 as a resin-bound intermediate to the pentamannoside 2.93, is reported in Fig. 2.33. In this scheme, the resin-bound linker 2.108 was placed in a sealed glass tube (Teflon stopper) presaturated with dry nitrogen and the donor 2.109 (3 eq) was injected as a dry DCM solution. The reaction was shaken at rt for 15 min, then trimethylsilyl trifluoromethane sulfonate (TMSOTf, 0.5 M in DCM, 0.3 eq) was added and the vessel was stirred at rt for 1 h. The solution was then removed under nitrogen and several washing cycles with dry DCM–dry THF removed the soluble impurities. After drying the resin in vacuo, a small aliquot of the resin-bound protected saccharide 2.90 (1–2 mg) was cleaved (dry silver triflate, DCM–MeOH 10/1, ultrasound mixing, 10 min), mixed with a matrix solution and directly submitted to MALDI–TOF MS in the positive-ion mode to check the purity of the intermediate, which resulted to be excellent. Compound 2.90 was deacetylated with 0.5 M NaOMe–MeOH (6 eq) in DCM–MeOH (10/1) as a solvent with stirring at rt for 2 h. The solution was then removed and multiple washings with crown ether–AcOH in THF removed any trace of the deacetylation reagent. The usual DCM–THF washing cycles and drying in vacuo produced the resin-bound, deacetylated 2.91. The same coupling–purity check cycle was repeated and the resin-bound, acetylated disaccharide 2.92 was finally dissolved into acetone–water 9/1 and oxidatively cleaved (NBS, DTBP, 4 eq) for 90 min at rt. The solution was recovered together with resin washings (THF–DCM), then triethylsilane was added to destroy the unreacted NBS. The solution was azeotroped with toluene and the residue was chromatographed (silicagel flash, then RP18 (reverse phase) flash) to give pure 2.106 with a 75% yield calculated on the resin loading of linker 2.108 (Fig. 2.33).
|
|
|
|
|
|
Ac |
|
|
|
|
a |
BnO |
O |
|
|
|
|
|
|
O |
|
||
|
O |
SH |
|
BnO |
|
|
|
|
|
BnO |
|
|
|
||
|
|
|
|
|
|
||
|
2.108 |
|
|
|
2.90 |
S |
|
|
|
|
OH |
|
|
||
|
|
BnO |
|
|
O |
|
|
|
b |
BnO |
O |
|
|
|
|
|
|
BnO |
|
|
|
|
|
|
|
|
2.91 S |
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
Ac |
|
|
|
|
Ac |
|
BnO |
O |
|
|
|
BnO |
O |
|
BnO |
O |
|
|
|
BnO |
O |
a |
BnO |
|
|
|
c |
BnO |
|
|
BnO |
O |
|
|
|
BnO |
O |
|
BnO |
O |
|
|
|
BnO |
O |
|
BnO |
|
2.92 |
|
|
BnO |
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
|
2.106 |
OH |
|
|
|
O |
|
|
75% from 2.108 |
|
a:2.109, TMSOTf, DCM, rt, 1 hr; b: 0.5M NaOMe, DCM/MeOH 10/1, rt, 2 hrs;
c:NBS, DTBP, acetone/water 9/1, rt, 90'.
Figure 2.33 SP synthesis of compound 2.106.