

методичка фкх
.pdfсупроводжується асиміляцією електронів, наприклад Сu2++2е=Сu, на другому (аноді) – окислювальний, наприклад Zn–2e=Zn2+, що є джерелом електронів.
Часто гальванічні елементи записують схематично, при цьому показують послідовність контактів реагуючих речовин, які перебувають в різних фазах, розділяючи їх вертикальними лініями. Наприклад, запис елемента ДаніеляЯкобі
(–)Cu/Zn/ZnSO4//CuSO4/Cu(+)
Спостерігаємо три міжфазових стрибки електричного потенціалу:
Cu/Zn, Zn / Zn2 , Cu 2 / Cu
які свідчать, що елемент Даніеля-Якобі є нерівноважною електрохімічною системою, в якій виникає різниця потенціалів Е, що дорівнює сумі міжфазових стрибків потенціалів:
Å Cu/Zn Zn/Zn2 Cu2 / Cu |
(5.1) |
де Е – електрорушійна сила (ЕРС) гальванічного елемента.
Оскільки чисельні значення міжфазових потенціалів не піддаються експериментальному визначенню, в електрохімічній практиці для розрахунків ЕРС гальванічних елементів широко використовують рівняння, що зв’язує ЕРС з електродними потенціалами .
Тоді ЕРС дорівнює максимальній різниці електродних потенціалів:
Å Cu2 / Cu |
- Zn/Zn2 |
(5.2) |
За визначенням електродний потенціал будь-якого напівелемента – це ЕРС ланцюга, який складено з на півелемента, що досліджується і напівелемента порівняння, електродний потенціал якого постійний. Таким електродом порівняння в електрохімії може бути нормальний водневий електрод (НВЕ), потенціал якого прийнятий рівним нулю.
В гальванічному елементі Даніеля-Якобі відбувається окислення цинку і перехід в розчин його іонів
Ζn – 2е Ζn2+
і відновлення іонів міді та осадження їх на електроді Сu2++2е Сu.
Цинковий електрод в даному елементі буде мати від’ємний заряд, а мідний – додатній. Електрони переходять по замкнутому металевому провіднику від цинкового електроду до мідного.
Сумарна реакція в елементі становить:
Ζn+ Сu2+ Ζn2++ Сu.
Якщо процес, в гальванічному елементі провести в зворотних умовах, за Р=const і T=const, то в елементі виконує максимальна корисна робота, що дорівнює зменшенню ізобарного потенціалу системи:
А’max=– GT=ZFE |
(5.3) |
де Z – число електронів, що беруть участь в реакції; F – стала Фарадея, 96493; E – ЕРС гальванічного елемента.
Для цієї реакції запишемо рівняння ізотерми:
31

G R T ln Kc R T ln |
aZn2 aCu |
------ |
(5.4) |
|||
|
||||||
|
a |
Cu |
2 a |
Zn |
|
|
|
|
|
|
|
||
де активності нейтральних атомів цинку та міді дорівнюють: aZn=аCu=1. Виходячи із рівняння (5.3)
Z F E R T ln KC |
R T ln |
аZn2 |
(5.5) |
||||||||||
a |
Cu |
2 |
|||||||||||
|
|
|
|
|
|
|
|
||||||
звідки, |
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
aCu2 |
|
|
|
|
|||||
Е |
R T |
ln KC |
R T |
ln |
|
. |
(5.6) |
||||||
|
|
|
|
||||||||||
|
Z F |
Z F а |
Zn |
2 |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|||
Якщо активності всіх речовин реакції дорівнюють одиниці, то ЕРС (Е) приймає значення стандартної ЕРС (Ео)
Ео ZR TF ln KC ,
де Ео– стандартна ЕРС.
Таким чином, рівняння (5.6) можна записати в загальному вигляді:
Е Ео |
R T |
ln aокис . |
|
||
|
Z F авідн |
|
По аналогії для електродного потенціалу одержимо
о ZR TF ln амеZn ,
(5.7)
(5.8)
(5.9)
де о – нормальний електродний потенціал, що дорівнює потенціалу рівноважного оберненого електрода, зaнуреного в розчин, в якому активність потенціал визначаючих іонів дорівнює одиниці. Співвідношення (5.8) і (5.9) мають назву – рівняння Нернста–Тюріна.
Стандартну ЕРС можна визначити як різницю електродних потенціалів:
Ео кo ao . |
(5.10) |
Нормальні електродні потенціали різних металів виміряні відносно нормального водневого електрода і зведені в таблицю. Якщо розмістити метали в порядку зростання їх нормальних електродних потенціалів, отримаємо так званий ряд напруги.
Всі метали, розміщені в ряду напруг до водню, мають негативний нормальний електродний потенціал, тобто на таких електродах при роботі відповідного гальванічного елемента відбувається процес окиснення металу. Позитивний нормальний електродний потенціал, який мають метали, які стоять за воднем, означає, що відносно нормального водневого електроду даний електрод заряджується позитивно, тобто при роботі елемента відбувається процес відновлення металу.
Електроди першого роду – це металеві або газові електроди, занурені в розчин своїх іонів і обернені відносно цих іонів (аніонів і катіонів), від активності яких залежить потенціал електроду. Прикладом електродів першого роду можуть бути мідний Cu/Сu2+ , цинковий Zn/Zn2+ , водневий
Pt(H2)H+, хлорний Pt(Cl2)Cl– тощо.
32

Нормальний водневий електрод (рис. 5.3) складається із платини, покритої платиновою черню і частково зануреної в розчин, в якому активність іонів водню дорівнює одиниці, а тиск водню в газовій фазі –101325 Па. Потенціал такого електрода умовно беруть рівним нулю.
Електродна реакція, що проходить на платині:
|
|
|
|
|
|
|
|
|
|
1/2Н2 Н++е |
|
|||||
|
|
|
|
Потенціал водневого |
електрода |
|||||||||||
|
в |
|
загальному |
випадку |
визначається |
|||||||||||
|
рівнянням |
|
|
|
|
aH |
|
|
|
|||||||
|
|
|
|
|
о |
|
|
|
|
R T |
ln |
, |
|
(5.11) |
||
|
Н |
/ Н |
/ |
Н |
|
P1/ 2 |
|
|||||||||
|
|
|
Н |
|
|
F |
|
|
|
|||||||
Рис. 5.3. Нормальний водневий |
|
|
|
|
|
|
|
|
|
|
|
H2 |
|
|
|
|
де |
|
|
Но / Н – |
|
нормальний |
потенціал, |
||||||||||
електрод: 1 – посудина; 2 – платиновий |
|
|
|
|||||||||||||
електрод; 3 – скляна трубка; 4, 8 – крани; 5 – |
прийнятий |
|
рівним |
нулю; |
P – |
|||||||||||
трубка введення водню; 6 – гідрозатвір; 7 – |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H2 |
|
рівноважний |
тиск |
водню |
над |
|||||||||||||
сифон. |
||||||||||||||||
платиною.
Оскільки вимірювання з водневим електродом проводять приблизно при тиску 1 атм
|
|
|
|
|
R T |
ln a |
|
, |
(5.12) |
|
|
|
|
||||||
|
Н |
|
/ Н |
|
F |
H |
|
|
|
|
|
|
|
|
|
|
|||
Незручності при користуванні водневим електродом обумовлені необхідністю мати очищений водень, який безперервно пропускають через розчин. Потенціал водневого електрода нестійкий і встановлюється повільно, тому на практиці як електрод порівняння використовують електроди іншого роду: каломельний, хлорсрібний тощо.
Електроди другого роду – це метали, вкриті шаром малорозчинної солі металу і занурені в розчин добре розчинної солі, яка має спільний аніон з малорозчинною сіллю. Ці електроди можна розглядати як зворотні відносно аніона, тому їх потенціал можна розрахувати через активність аніонів. Прикладом електродів другого роду є каломельний, хлоросрібний тощо.
33
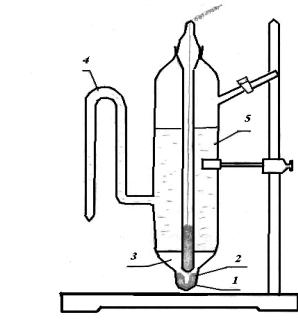
|
Каломельний електрод (рис. 2.4) |
|||||
|
складається |
з |
ртуті, |
пасти, |
||
|
виготовленої |
з |
|
каломелі, |
||
|
платинового дроту, мідного дроту- |
|||||
|
виводу, |
пробки і сифону. |
Електрод |
|||
|
заливають розчином |
КСl, |
залежно |
|||
|
від концентрації якого розрізняють |
|||||
|
0,1н, нормальний (1,0н КСl) і |
|||||
|
насичений каломельні електроди. |
|||||
|
В |
каломельному |
електроді |
|||
|
проходить реакція |
|
|
|||
|
|
|
|
Hg–e Hg+. |
|
|
|
Електродний потенціал |
|
||||
|
дорівнює: |
|
|
|
||
|
кал кало |
|
R T |
ln аСl |
(5.13) |
|
Рис. 2.4. Каломельний електрод: 1 – |
F |
|
||||
|
|
|
|
|
||
ртуть: 2 – платиновий дріт; 3 – каломель; 4 – сифон; 5 – розчин KCl.
Окислювально-відновні електроди – це електроди, матеріал яких не бере участі в електродній реакції, а тільки в передачі електронів від окислювача до відновлювача, що перебувають в одному розчині. Прикладом такого електрода може бути платина, занурена в розчин, що містить Fe2+ і Fe3+ або Sn2+ і Sn4+. Відмінність окислювально-відновних електродів від інших полягає в тому, що продукти окислення або відновлення залишаються в розчині, а не виділяються на електродах.
Електродний потенціал такого електрода, |
наприклад Pt/Sn4+/Sn2+ |
|
||||||||||||
|
|
4 |
|
2 |
î |
4 |
|
2 |
R T |
ln |
aSn4 |
|
, |
(5.14) |
|
|
|
2 F |
aSn2 |
|
|||||||||
|
Sn |
|
/ Sn |
|
Sn |
|
/ Sn |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
||||
Серед Red-Oxy-електродів широко застосовується хінгідронний електрод, що використовується для визначення концентрації водневих іонів в розчині. Це платиновий електрод, занурений в розчин хінгідрону. Хінгідрон (С6Н4)2O2(OН)2, розпадається у водному розчині на еквівалентні кількості хінону С6Н4O2 і гідрохінону С6Н4(OН)2. Останній є двохосновною кислотою, дисоціює з утворенням аніона С6Н4O2–, який при окисленні переходить в хінон. Сумарно реакцію можна виразити рівнянням
С6Н4(OН)2 С6Н4O2+2Н++2е
Потенціал хінгідронного електрода
xr |
xrî |
0,059 lg aH , |
(5.15) |
тобто потенціал хінгідронного електрода визначається концентрацією іонів водню або рН розчину.
Вимірювання електродних потенціалів окремих електродів (напівелементів) проводиться лише у формі гальванічних елементів, складених із напівелемента, електродний потенціал якого належить
34
визначенню і стандартного електрода порівняння каломельного, електродний потенціал якого відносно нормального водневого електрода відомий.
Еокал=+0,2483 В.
Наприклад, для визначення потенціалів мідного і цинкового електродів в елементі Даніеля-Якобі з допомогою каломельного електрода порівняння складають два ланцюги
(–)Hg/Hg2Cl2/KClнас//CuSO4/Cu(+)
(–)Zn/ZnSO4//KClнас/Hg2Cl2/Hg(+)
і визначають їх ЕРС.
В першому колі каломельний електрод грає роль негативного електрода, в другому – позитивного. Знаки електродів визначають, користуючись таблицею стандартних електродних потенціалів і порівнюють їх з потенціалом електрода
порівняння. У вказаних елементах проходять такі електрохімічні реакції: 2Cl–+2Hg–2е→ Hg2Cl2 ; Cu2++2е→ Cu
Zn–2е→Zn2+ ; Hg2Cl2+2е→2Hg+2Cl–
Значення потенціалів визначають на підставі таких міркувань, знаючи ЕРС гальванічного елемента:
ЕCu-кал= Сu – кал, |
(5.15) |
потенціал катоду дорівнює |
|
Сu=ЕCu-кал + кал. |
(5.16) |
Маючи виміряну ЕРС гальванічного елемента |
|
ЕZn-кал= кал – Zn, |
(5.17) |
потенціал аноду дорівнює |
|
Zn= кал – ЕZn-кал. |
(5.18) |
Отримані значення електродних потенціалів порівнюють з табличними або розрахованими за рівнянням Нернста-Тюріна.
Завдання на виконання роботи
1.Вимірюємо ЕРС гальванічних елементів, складених із електродів першого та другого роду.
2.Використовуючи одержані результати розрахувати окремі електродні потенціали.
Прилади, лабораторний посуд та реактиви
Мультиметр, електроди першого роду, каломельний електрод, насичений розчин КСІ, електричні провідники.
Порядок виконання роботи
1.Складають гальванічний ланцюг, використовуючи два напівелементи з електродів першого роду, на прикладі елементу Даніеля-Якобі, підключають його до мультиметра і вимірюють ЕРС.
2.Записують електрохімічну схему досліджуваного гальванічного елемента та рівняння окисно-відновної реакції, що проходить під час роботи даного гальванічного елемента.
35
3.Складають гальванічні ланцюги, використовуючи один із досліджуваних напівелементів та електрод порівняння (каломельний), підключають їх до мультиметра і вимірюють їх ЕРС.
4.Записують електрохімічну схему досліджуваних гальванічних елементів та рівняння окисно-відновних реакцій, що проходить під час роботи даного гальванічного елемента.
5.Використовуючи рівняння 5.15, 5.16, 5.17 та 5.18 розраховують електродні потенціали досліджуваних напівелементів.
6.Розраховують ЕРС досліджуваного елемента, використовуючи розраховані значення електродних потенціалів.
7.Оцінюють похибку експеременту, порівнюючи виміряне значення ЕРС гальванічного елементу (п.1) та розраховане значення ЕРС (п.6).
Питання для самоперевірки
1.Причини виникнення ЕРС у гальванічних елементах (ГЕ).
2.Від яких факторів залежить значення електродного потенціалу? Що таке нормальний електродний потенціал?
3.Що таке ряд напруг?
4.Які рівняння передають взаємозв’язок ЕРС гальванічного елемента з максимальною роботою та ізобарним потенціалом?
5.Як класифікують електроди? Дайте визначення електроду першого роду, електроду другого роду та окисно-відновного електроду.
6.Які електроди використовують в якості електродів порівняння?
7.Наведіть реакції на електродах залежно від потенціалу електрода, що працює в парі.
8.Як вимірюють експериментально значення електродних потенціалів?
9.Навести рівняння Нернста-Тюріна для розрахунку електродних потенціалів та ЕРС гальванічних елементів.
Лабораторна робота №6 ХІМІЧНАКІНЕТИКА
Мета роботи: вивчення кінетики реакції гідролізу сахарози та визначення константи швидкості процесу розчинення бензойної кислоти у воді.
Теоретичні відомості
Хімічна кінетика вивчає швидкість проходження хімічних реакцій та залежність її від різноманітних факторів – концентрації реагуючих речовин, температури, впливу каталізаторів тощо.
Прості речовини та хімічні сполуки, що вступають до реакції, називаються вихідними речовинами. Кінцеві речовини, які утворюються в реакції і не зазнають змін в ході перебігу подальших хімічних перетворень, називаються продуктами реакції. Речовини, що виникають в одних і зникають в
36
інших стадіях хімічного процесу, називаються проміжними, а реакції утворення та розкладу цих речовин – проміжними.
Розрізняють два типи реакцій: гомогенні та гетерогенні. Гомогенними називають такі реакції, при яких всі вихідні речовини і продукти реакції перебувають в одній і тій самій фазі, наприклад, в газовій, або в розчині. Гетерогенними називають такі реакції, при яких процес відбувається на межі розділу двох фаз, і речовини, що беруть участь у реакції (в тому числі і каталізатор), перебувають у різних фазах.
Швидкістю хімічної реакції називають зміну концентрації будь-якої з речовин, що беруть участь у реакції за одиницю часу і в одиниці певного об’єму. Швидкість реакції знаходять, як правило, за концентрацією вихідних речовин. Позначивши через υ швидкість реакції, що проходить за постійної температури, dС – зміну концентрації однієї з вихідних речовин за
нескінченно малий час реакції dτ, одержуємо |
|
dC |
(6.1) |
d |
|
Знак "–" у рівнянні (6.1) береться тому, що позитивному приросту часу відповідає негативна зміна концентрації; а за фізичним змістом швидкість реакції завжди позитивна.
В основі хімічної кінетики одностадійних реакцій лежить закон дії мас, згідно з яким швидкість хімічної реакції пропорційна добутку концентрацій реагуючих речовин. Причому кожна з концентрацій береться в степені, що дорівнює стехіометричному коефіцієнту (п1, п2, п3,...), який стоїть перед формулою даної сполуки в рівнянні реакції.
На підставі цього закону можна записати:
|
dC |
n |
n |
|
n |
|
(6.2) |
|
d |
K C1 1 |
C2 |
2 |
C3 |
3 |
Коефіцієнт пропорційності К для даної реакції при сталій температурі є величиною постійною і називається константою швидкості реакції. На відміну від швидкості υ константа швидкості реакції К не залежить від концентрації для даної реакції при даній температурі. З рівняння (6.2) випливає фізична суть константи швидкості: вона чисельно дорівнює швидкості реакції за умови, що концентрація кожної з вихідних речовин дорівнює одиниці C1=C2 =С3 =1
Кінетично хімічні реакції розрізняють за ознакою молекулярності або порядку реакції.
Молекулярність реакції визначається числом молекул, одночасною взаємодією яких здійснюється елементарний акт хімічного перетворення. Розрізняють реакції мономолекулярні, бімолекулярні, тримолекулярні. (Останні трапляються надзвичайно рідко).
Молекулярність реакції не відбиває наявності проміжних стадій. Швидкість складної реакції визначається швидкістю найповільнішої стадії, тобто кінетично складна реакція описується закономірностями цієї стадії. У зв’язку з цим складні реакції зручніше характеризувати не молекулярністю, а порядком. Порядок реакції дорівнює сумі показників ступенів концентрацій у
37
рівнянні, яке передає залежність швидкості реакції від концентрації реагуючих речовин.
Для реакцій першого порядку кінетичне рівняння включає концентрацію однієї речовини:
dC |
K C , |
(6.3) |
d |
|
|
Розділяючи змінні та інтегруючи рівняння (6.3), одержуємо
dC |
K d ;ln C K const , |
(6.4) |
С |
|
|
Сталу інтегрування визначають, беручи до уваги, що при τ=0 і С=Со (Со– початкова концентрація), lnСо = const. Підставляючи це значення у рівняння (6.4), одержуємо кінетичне рівняння реакції першого порядку:
K |
1 ln |
Co |
(6.5) |
|
C |
||||
|
|
|
Якщо початкову концентрацію позначити а (тобто Cо), а кількість речовини, яка зазнала змін за час τ, через х (тобто C = а – x),одержуємо
K |
1 ln |
a |
(6.6) |
|
a x |
||||
|
|
|
Одна з важливих кінетичних характеристик – період піврозпаду τ1/2 – час, за який прореагує половина початкової кількості речовини, тобто момент, коли х=а/2.
Підставивши це значення у рівняння (6.6), одержуємо період піврозпаду для реакції першого порядку
1/ 2 |
|
ln 2 |
. |
(6.7) |
|
||||
|
|
K |
|
|
Отже, період піврозпаду для реакцій першого порядку не залежить від початкової концентрації.
Якщо один з компонентів міститься у великому надлишку, тобто його концентрація практично не змінюється з часом, деякі бімолекулярні реакції протікають за законами реакцій першого порядку. Класичним прикладом такої реакції є реакція гідролізу (інверсії) сахарози у водному розчині або розчинення бензойної кислоти у воді. Такі реакції називаються
псевдомономолекулярними.
Порядок реакції встановлюється експериментально. Один з простих методів полягає в тому, що дослідні дані зміни концентрації з часом підставляють в кінетичне рівняння першого або другого порядків. Порядок рівняння визначають за рівнянням, яке найкраще задовольняє умову сталості константи швидкості. Якщо період піврозпаду не залежить, від початкової концентрації, говорять про реакцію першого порядку; якщо він обернено пропорційний початковій концентрації – про реакцію другого порядку.
При підвищенні температури швидкість хімічних реакцій зростає. Для більшості реакцій справджується приблизне правило Вант-Гоффа, згідно з яким при підвищенні температури на 10 °С збільшується константа швидкості реакції в 2–4 рази. Математично це можна записати так,
38
Kt 10 |
2...4. |
(6.8) |
Kt |
|
|
Більш точно залежність константи швидкості реакції від температури Т можна пояснити, використавши рівняння Арреніуса:
d ln K |
|
E |
, |
(6.9) |
|
dT |
R T |
||||
|
|
|
де Е – енергія активації; R – універсальна газова стала. Після інтегрування рівняння (6.9) набуває вигляду
ln K |
E |
const, |
(6.10) |
|
R T |
||||
|
|
|
або представивши в експоненційній формі, отримаємо:
K Ko e E / R T , |
(6.11) |
де Ko – емпірична константа, яка називається передекспоненційним множником.
Розглянемо фізичний зміст поняття "енергія активації". Умовою протікання реакції є ефективне зіткнення часток, число яких становить малу частку від загального числа зіткнень. Ефективними є зіткнення між активними часточками, які мають підвищений запас енергії. Енергія активації – це мінімальна енергія, яку повинні мати реагуючі часточки, щоб зіткнення між ними призвело до реакції. Частки, енергія яких більше або дорівнює Е, називаються активними. Активацію можна здійснити, підводячи до системи енергію, за рахунок нагрівання системи, або здійснюючи електричний розряд опроміненням. Енергію активації визначають експериментально.
Каталізаторами називають речовини, які, як правило, прискорюють хімічну реакцію, беруть участь в тій чи іншій стадії реакції, але в результаті реакції залишаються незмінними хімічно.
Збільшення швидкості реакції під дією каталізатора можна пояснити тим, що каталізатор знижує енергію активації реакції, збільшуючи тим самим частину активних молекул. Одночасно каталізатор збільшує ентропію активації.
За агрегатним станом каталізатора і речовин, які беруть участь у реакції, каталіз поділяють на гомогенний і гетерогенний.
У випадку гетерогенного каталізу хімічна реакція проходить на межі розділу фаз, каталізатор – реагуючі речовини.
При гомогенному каталізу каталізатори перебувають в одній фазі з реагуючими речовинами. Найпоширеніші гомогенні каталізатори – кислоти, основи, іони перехідних металів та їх комплекси і біологічні каталізатори (ферменти або ензими).
Каталітичним реакціям притаманні такі особливості:
–процесі реакції кількість каталізатора залишається незмінною;
–каталізатор не змінює хімічних властивостей, але може змінюватись фізично – старіти, "отруюватись";
39
–мізерно мала кількість каталізатора порівняно з кількістю реагуючої речовини значно змінює швидкість реакції;
–як правило, дія каталізатора специфічна, але існують каталізатори, які прискорюють багато реакцій, наприклад, іони Н+ у гомогенному каталізі;
–в оберненій реакції каталізатор не зміщує рівноваги, однаковою мірою прискорюючи пряму та зворотну реакції.
На реакції, термодинамічно неможливі, каталізатор не впливає.
Одним з найпоширеніших видів каталізу є кислотно-основний. Типовий приклад кислотного гомогенного каталізу – реакція гідролізу сахарози в присутності кислоти. Згідно з теорією протолітичної рівноваги, кислота в процесі реакції віддає протон (донор протонів). Іонізація кислот
відбувається внаслідок їх протолітичної реакції з розчинником, в результаті чого з’являється іон гідроксонія Н3О+ – каталітично діюче начало у кислотному каталізі. Наприклад, для HCl у воді НСІ+Н2О=Cl–+H3O+.
При кислотному каталізі нa першій стадії відбувається протолітична реакція і протонізація вихідної речовини за раxунок передачі протона від каталізатора (кислоти). Друга стадія – саме хімічне перетворення з утворенням продуктів реакції і відщепленням протона. При цьому каталізатор (кислота) регенерується. Лімітуючою стадією сумарної двостадійної реакції буде найповільніша стадія. При кислотному каталізу реакції гідролізу сахарози лімітуючою виявляється друга стадія.
С12Н22О11+Н3О+ С12Н22О11Н++Н2О (І стадія) сахароза
С12Н22О11Н++2Н2О С6Н12О6+ С6Н12О6+ Н3О+ (ІІ стадія) фруктоза глюкоза
Друга стадія проходить за рівнянням кінетики реакції першого порядку, бо
вода присутня у великому надлишку і швидкість υ реакції становить |
(6.12) |
K Cсах Н , |
де К – істинна стала швидкості кислотно-каталітичного перетворення.
Угетерогенному процесі взаємодія відбувається на поверхні розділу фаз або в безпосередній близькості від неї. У системах з двох або більше компонентів взаємодія на поверхні розділу фаз призводить до виникнення різниці у складах поверхневого і внутрішнього шарів даної фази і, отже, процесу вирівнювання складів цих шарів. Якщо процес не прискорюється перемішуванням, а відбувається лише внаслідок дифузії, швидкість всього процесу, як правило, визначається швидкістю вирівнювання складів (швидкість найповільнішої стадії). На поверхні розділу фаз рівновага досягається значно скоріше. Прикладом гетерогенного процесу може бути процес розчинення бензойної кислоти у воді.
Убільшості гетерогенних процесів спільна швидкість дуже збільшується при перемішуванні. Але вплив дифузії в цьому випадку не зникає, бо при перемішуванні вирівнюється концентрація в більшій частині об’єму, але біля самої поверхні розділу завжди залишається
40