

Multiple Bonds Between Metal Atoms / 08-Rhenium Compounds
.pdfRhenium Compounds 291
Walton
in 8.5. The Re–Re bond length of 2.4705(5) Å is shorter by c. 0.07 Å than that in 8.5. Ab initio SCF and CI studies on the model species O3ReReCl2(H2PCH2PH2)2 have been interpreted140 in terms of a charge distribution Re(V)–Re(III) (i.e. d2–d4), with the short, strong Re–Re bond represented in terms of a μ-donation from [ReO3]- to [ReCl2(H2PCH2PH2)2]+
8.5 |
8.6 |
Fig. 8.6. The structure of the “dimer of dimers” molecule [(PMe3)3ClRe(µ-O2CPh)Re(O)]2(µ-O)2.
While Re2(O2CCH3)2Cl4L2 (L = H2O or py) react with PMe3, PMe2Ph and PMePh2 in alcohol solvents (ROH) to give the Re24+ complexes Re2Cl4(PR3)4 (Section 8.5.4), a quite different reaction course ensues with the phosphine ligand PPh3 and other triarylphosphines.98,141-143 Upon reacting cis-Re2(O2CCH3)2Cl4(H2O)2 in methanol with PAr3 ligands that have relatively low basicities (pKa values of 1.0-4.6) and moderately large cone angles (145-165°) the unsymmetrical Re26+ methoxides (MeO)2Cl2ReReCl2(PAr3)2 (PAr3 = PPh3, P(p-tolyl)3, P(m-tolyl)3, P(p-ClPh)3 and P(p-MeOPh)3) are formed.98,141-143 In the case of PPh3 this same type of product has been obtained for the bromide and with other alkoxide ligands, i.e. Re2X4(OR)2(PPh3)2 (X = Cl or Br; R = CH3, C2H5, n-C3H7 or i-C3H7).98,141 The reaction of cis-Re2(O2CCH3)2Cl4(H2O)2 with P(p-MeOPh)3 in methanol have also been found142 to produce the tetranuclear complex Re2(µ-O)4Cl4[P(p-MeOPh)3]4. The mixed halide-alkoxide products are different structurally from the Re(III)–Re(III) derivatives Re2X6(PR3)2 (see Sec. 8.4.4) since they possess the unsymmetrical mixed-valence Re(IV)–Re(II) structure shown in Fig. 8.7. Both Re2Cl4(OEt)2(PPh3)2 and Re2Cl4(OMe)2[P(p-MeOPh)3]2 have been crystallographically characterized; the Re–Re distances are 2.231(1) Å and 2.2476(4) Å, respectively.98,141,143 The very short Re–Re distance and eclipsed rotational geometry are in accord with the retention of a Re–Re quadruple bond, with one component of this bond being dative in character in the sense Re(1) Α Re(2), i.e. Re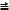 Re. These are interesting and relatively rare examples of intramolecular disproportionation reactions that occur at a metal-metal multiple bond without change in the formal metal-metal bond order. Indeed, they were the first examples of their kind to be reported.98,141 In the case of
Re. These are interesting and relatively rare examples of intramolecular disproportionation reactions that occur at a metal-metal multiple bond without change in the formal metal-metal bond order. Indeed, they were the first examples of their kind to be reported.98,141 In the case of
292Multiple Bonds Between Metal Atoms Chapter 8
the more basic phosphines PCyPh2 and PBz3, which have cone angles of 153° and 165°, respectively, their reactions with cis-Re2(O2CCH3)2Cl4(H2O)2 in methanol afford Re2(µ-O2CCH3)- Cl3(OMe)2(PCyPh2)2 (see 8.7), which has a slightly longer Re–Re quadruple bond distance because of the presence of an axial Re–Cl bond (see Table 8.1), and the paramagnetic Re25+ complex Re2(µ-O2CCH3)Cl4(PBz3)2 (see Section 8.5.4), respectively. The tris-ethoxide complex Re2Cl3(OEt)3(PPh3)2, which has a structure similar to that of Re2Cl4(OEt)2(PPh3)2 but with one Re–Cl bond replaced by Re–OEt (see 8.8) is formed by the reactions of Re2Cl4(OEt)2(PPh3)2 or Re2Cl6(PPh3)2 (Section 8.4.4) with NaOEt in ethanol.143
Finally, note should be made of some screening studies that have been carried out involving the use of [Re2(O2CC2H5)4]SO4 and several derivatives of the type cis-Re2(O2CR)2X4(H2O)2 as anti-tumor agents.144
8.7 |
8.8 |
Fig. 8.7. The structure of Re2Cl4(OMe)2[P(p-MeOPh)3]2, an example of an intramolecular disproportionation product.
8.4.3 Other anionic ligands
In addition to substitution reactions that involve monodentate anionic ligands (Section 8.4.1) and bridging carboxylate ligands (Section 8.4.2) there is also an extensive body of literature that deals with the reactions of the [Re2X8]2- anions, and in some cases carboxylate complexes of the types Re2(O2CR)4Cl2 and Re2(O2CR)2Cl4L2, with bidentate monoanionic and dianionic ligands. In most instances such ligands bridge the dirhenium unit, but in a few cases chelation is found to occur. Examples of the latter are encountered in the formation of Re2X4(acac)2 and Re2X4(acac)2L2 (X = Cl or Br; L = DMSO, DMF or acacH) by the reactions of (NH4)2Re2X8·2H2O with acetylacetone.145-147 The structures of the DMSO and acetylacetone adducts Re2Cl4(acac)2L2 have been determined145,146 (see 8.9) and the presence of a quadruple bond confirmed (Table 8.1). The axial Re–O distance involving the neutral acacH ligand is very long (2.63 Å); this compares to distances of 2.01-2.02 Å for the chelating equatorial acac

Rhenium Compounds 293
Walton
anions.146 Another case, albeit an unusual one, is found in the mixed-metal quadruply bonded porphyrin complex [(TPP)MoRe(OEP)]PF6.148 In the dimetal cation the Mo–Re distance is 2.236 Å and the porphyrin ligands are perfectly eclipsed. This is one of several of several heterodinuclear complexes with multiple metal-metal bonds.149
8.9
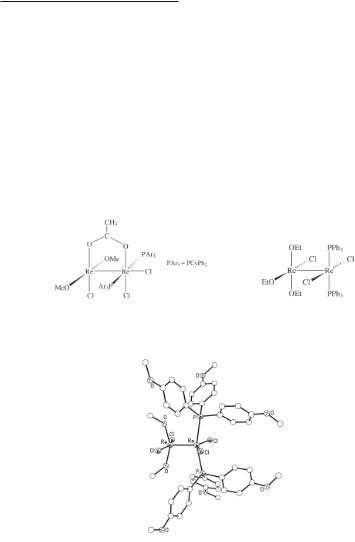
The reactions of (Bu4N)2Re2Cl8 with sulfate/sulfuric acid mixtures and with phosphoric acid have been shown to produce complexes whose structures are of the ‘acetate type’,150-152 in which the anionic ligands bridge the two metal atoms. These results further support the idea that substitution of some or all of the halide ligands of [Re2X8]2- usually leads to products in which the Re–Re quadruple bond (Table 8.1) is retained. The tetrakis(sulfato) derivative (NH4)2Re2(SO4)4(H2O)2 has also been prepared by the reaction of conc H2SO4 with various dirhenium(III) formate complexes.85 The structure of the [Re2(SO4)4(H2O)2]2- anion is shown in Fig. 8.8 and reveals the presence of two weakly bound water molecules (r(Re–O) = 2.28 Å). Like the carboxylate complexes Re2(O2CR)4Cl2, the sulfate Na2Re2(SO4)4·8H2O may be reconverted to (Bu4N)2Re2Cl8 upon reaction with refluxing hydrochloric acid in the presence of Bu4NCl.150 A compound that is closely related to this sulfate complex is formed upon reacting (Bu4N)2Re2Cl8 with phosphoric acid in methanol. Addition of CsCl to the resulting reaction mixture affords pale-blue crystalline Cs2[Re2(HPO4)4(H2O)2], whereas the use of pyridine in place of CsCl gives the anhydrous pyridinium salt (pyH)2Re2(HPO4)4.152 The crystal structure of the closely related derivative Cs2[Re2(HPO4)4(H3PO4)2] has been determined by Koz’min and co-workers.153 Its preparation, which was different from that used to obtain Cs2[Re2(HPO4)4(H2O)2], involves the high pressure reduction of KReO4 in a 2:1 mixture of H3PO4 and HCl at 330 °C, followed by the addition of (NH4)H2PO4 and CsCl to accelerate the crystallization of the complex. As expected, the two H3PO4 molecules are axially bound and the Re–Re bond length (2.224(1) Å) is similar to that of the sulfate complex (2.214(1) Å).
Fig. 8.8. The structure of the [Re2(SO4)4(H2O)2]2- anion present in Na2Re2(SO4)4·8H2O.

294Multiple Bonds Between Metal Atoms Chapter 8
The complex with 2-hydroxypyridine, Re2(hp)4Cl2,154 whose structure is represented in 8.10, is of importance since this type of ligand system has proved to be especially effective in stabilizing dimetal units. The Re–Re bond distance of 2.206(2) Å is about 0.03 Å shorter than that in the carboxylates of the type Re2(O2CR)4X2 (Table 8.1). This compound was first obtained by reacting (Bu4N)2Re2Cl8 with molten 2-hydroxypyridine. It has also been prepared by a method that can easily be adaptable to its bromo and iodo analogs, viz., the reaction of (Bu4N)2Re2X8 (X = Cl, Br or I) with Hhp in refluxing n-pentanol.155 The complexes Re2(hp)2Cl4(Hhp), and Re2(mhp)2X4(Hmhp)·S (X = Cl or Br; S (solvent of crystallization) = THF or (CH3)2CO) have also been prepared, but not from (Bu4N)2Re2X8; instead, the bis(acetate) complexes Re2(O2CCH3)2X4L2 (X = Cl or Br; L = H2O or 4-Mepy—see Section 8.4.2) were used as the starting materials with THF or acetone as the reaction solvent.136 The coordinated Hmhp molecule in Re2(mhp)2X4(Hmhp)·(CH3)2CO can be substituted by 4-methylpyridine to give Re2(mhp)2X4(4-Mepy).136 The reaction of Re2(hp)2Cl4(Hhp) with excess Hhp in hot ethanol affords the tetrakis derivative Re2(hp)4Cl2.136 The structure of Re2(mhp)2Cl4(Hmhp) is shown in Fig. 8.9 and reveals the presence of a trans arrangement of mhp ligands and an axially bound Hmhp ligand; the Re–Re distance of 2.210(1) Å is similar to that in trans-Re2(O2CCH3)2Cl4 (Section 8.4.2). The mhp ligands are bound in a polar fashion, i.e., they are orientated in the same direction so as to give a dimetal unit that has [ReCl2O2] and [ReCl2N2] units. Formally at least, the Re centers can be considered to differ in oxidation state, viz., Re(IV) and Re(II), respectively. However, the ability of the mhp ligands to delocalize charge makes these systems behave more like symmetrical quadruply bonded Re(III)–Re(III) species than mixed-valent Re(IV)–Re(II) derivatives. The mixed carboxylate-mhp species Re2(O2CR)(mhp)2X3 (R = CH3 or C2H5; X = Cl or Br) are also known;136 their chemistry is considered elsewhere (see Section 8.4.2). Another complex that contains a 2-hydroxypyridine ligand is Re2(µ-chp)2(δ2-chp)Cl3, which is formed from the reaction of Re2(O2CCH3)4Cl2 and molten 2-hydroxy-6-chloropyri- dine.156 It has a structure in which there are two trans head-to-head bridging chp ligands (i.e. similar to Re2(mhp)2Cl4(Hmhp)) and one chelating chp ligand with its nitrogen atom bound in an axial position. The Re–Re distance is normal (see Table 8.1).
Fig. 8.9. The structure of Re2(mhp)2Cl4(Hmhp).
The sulfur-containing analogs of Re2(µ-hp)4X2 are formed when 2-mercaptopyridine is reacted with (Bu4N)2Re2X8 (X = Cl or Br).157 The formation of Re2(µ-mp)4X2 probably occurs via the intermediacy of Re2(mp)2X4. The bis-acetate complexes cis-Re2(O2CCH3)2X4L2 (X = Cl

Rhenium Compounds 295
Walton
or Br; L = H2O or py) can be used as alternative starting materials to (Bu4N)2Re2X8.157 The structural characterization of Re2(µ-mp)4Cl2 shows that there is a small twisting about the Re–Re bond (torsion angles range from 0.8° to 11.8°) but the Re–Re quadruple bond distance is close to that in Re2(hp)4Cl2 (Table 8.1). However, what is surprising is that the stereoisomer that is formed is the one with a 3:1 orientation of the µ-mp ligands (8.11), while the cis 2:2 isomeric form is obtained in the case of Re2(hp)4Cl2 (8.10). There is no obvious reason for this difference, which may simply be the consequence of solubility differences between the different stereoisomers in the reaction solvents. Indeed, the structural characterization of the compound Re2(µ-C7H4NS2)4Cl2, which contains bridging N,S-benzothiazole-2-thiolate ligands, has shown that there is cis 2:2 orientation, like that in Re2(hp)4Cl2.158 In this molecule, there is a significant deviation from an eclipsed conformation such that the average torsion angle ρav is 18.0°. As a consequence, the Re–Re distance is a little longer (by c. 0.026 Å) than that in Re2(µ-mp)4Cl2. The synthetic procedure for obtaining Re2(µ-C7H4NS2)4Cl2 used Re2Cl6(PPh3)2 (see Section 8.4.4) rather than (Bu4N)2Re2Cl8.158
8.10 |
8.11 |
There is now a fairly extensive body of synthetic and structural data for dirhenium(III) complexes that contain bridging amidate, amidinate and related monoanionic ligands. The first of these to be reported was the bis-N,N'-diphenylbenzamidinato complex Re2[(PhN)2CPh]2Cl4 along with its mono-THF solvate, both of which have the expected ligand-bridged structure with a trans disposition of amidinate ligands.159 In the case of the solvated derivative, the THF molecule occupies one of the empty coordination sites colinear with the Re–Re bond. As a result, the Re–Re distance of 2.209(1) Å is longer than that in the complex lacking THF (2.177(1) Å). Other examples of structurally characterized amidinato-bridged complexes that were reported in earlier studies are Re2[(PhN)2CCH3]2Cl4 and Re2[(CH3N)2CPh]4Cl2 (Table 8.1).160 The structure of the first of these is very similar to that of Re2[(PhN)2CPh)]2Cl4, while the tetrakis-N,N'- dimethylbenzamidinato complex is noteworthy because the methyl groups keep the chloride ligands at a greater distance than that encountered in Re2(O2CR)4Cl2 compounds. As a result, the Re–Re distance in this amidinato complex is shorter (by 0.027 Å).160
The most thoroughly studied of the amidinate ligand systems are the diarylformamidinates [ArNC(H)NAr]- (abbreviated DArF). The first report on dirhenium(III) complexes that contain these bridges appeared in 1992 and involved the synthesis of the compound Re2(DTolF)4Cl2 by the reaction of molten di-p-tolylformamidine with Re2(O2CCH3)4Cl2.161 This complex can be reduced by Na/Hg to produce Re2(DTolF)4Cl and Re2(DTolF)4 (see Section 8.5.5) and substitution of the axial Re–Cl bonds by methoxide gives Re2(DTolF)4(OMe)2. Structural characterization of Re2(DTolF)4Cl2 and Re2(DTolF)4(OMe)2 shows that the Re–Re bond length in the methoxide is longer by c. 0.03 Å (Table 8.1). Subsequently, a variety of other complexes of the type Re2(DArF)4Cl2 have been prepared by a procedure similar to that used for Re2(DTolF)4Cl2, and the structures of several of them determined crystallographically (see Table 8.1).162 Extensive electrochemical characterizations have been carried on the series of compounds for which the aryl rings XC6H4 or X2C6H3 contains the following substituent(s) X: H, p-Me, p-MeO, m-MeO, p-Cl, m-Cl, p-CF3, m-CF3, 3,4-Cl2 and 3,5-Cl2.161,162 One of the axial

296Multiple Bonds Between Metal Atoms Chapter 8
Re–Cl bonds in Re2[(p-MeOC6H4N)2CH]4Cl2 can be replaced by BF4- to form the compound {Re2[(p-MeOC6H4N)2CH]4Cl}BF4 in which the Re–Re bond distance is shortened by c. 0.05 Å compared to its parent; a F atom of the BF4- anion is at a distance of 4.502 Å to the coordinatively unsaturated Re atom.163
Only one diarylformamidinate ligand has to date been found to form the pair of complexes Re2(DArF)2Cl4 and Re2(DArF)3Cl3, namely when Ar = phenyl.164,165 They resemble structurally the carboxylate-bridged complexes of these same types (vide supra). The compound Re2(DPhF)3Cl3 was prepared by the reactions of Re2Cl5(PMePh2)3 and Re2Cl4(PEt3)4 (see Section 8.5.4) with N,N'-diphenylformamidine, and converted into trans-Re2(DPhF)2Cl4 upon treatment with HBF4·Et2O in CH3CN/CH2Cl2.164 The mechanisms of these redox reactions are unknown. Later it was found that the compound trans-Re2(DPhF)2Cl4 could be prepared in good yield by a non-redox procedure involving the reaction of (Bu4N)2Re2Cl8 with the formamidine in molten state or in refluxing 1,2-dichlorobenzene.165 The compounds have been structurally characterized and the Re–Re distances follow the expected trends. This distance in Re2(DPhF)2Cl4(H2O)·2THF, which contains an axially bound H2O molecule, that is in turn H-bonded to two THF molecules (see Fig. 8.10), is longer (by c. 0.04 Å) than that in trans-Re2(DPhF)2Cl4.
Fig. 8.10. The structure of trans-Re2(DPhF)2Cl4(H2O)·2THF.
Other monoanionic bridging ligands that contain N,N donor atom sets include the anion of 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidine, which reacts as its Li+ salt with
(Bu4N)2Re2Cl8 to give Re2(µ-hpp)4Cl2 and Re2(µ-hpp)3Cl3, both of which have been structurally characterized (Table 8.1).166 The tetrakis-hpp complex was also prepared by the use of molten Hhpp. DFT calculations have been utilized to compare the electronic structures of Re2(HNCHNH)4Cl2 and Re2(hpp)4Cl2.166 The anions of N6,N6-dimethyladenine and 7-azain- dole have been used to prepare Re2(µ-dmad)4X2 (X = Cl or Br) (from Re2(O2CCH3)4X2) and Re2(µ-aza)4Cl2 (from (Bu4N)2Re2Cl8), which have been characterized by NMR spectroscopy.167 Different relative orientations of these unsymmetrical ligands about the Re–Re bond lead to mixtures of stereoisomers.
When Re2(O2CCH3)4Cl2 is reacted with molten amides, µ-amidato complexes of the type Re2[(µ-RNC(R')O]4Cl2 are formed.165,168 Attempts to isolate Re2[µ-RNC(R')O]2Cl4 compounds have not yet been successful.165 Most of the Re2[µ-RNC(R')O]4Cl2 complexes, all of which contain N,O ligand bridges, have been characterized by X-ray crystallography. Each Re atom contains cis or trans- ReN2O2 planar units and an axial Re–Cl bond; this cis or trans designation is used in Table 8.1 to differentiate these geometries. For the synthesis of Re2(µ-CyNCHO)4Cl2, the compound (Bu4N)2Re2Cl8 was used in place of Re2(O2CCH3)4Cl2.165 The lability of the terminal Re–Cl bonds in trans-Re2[µ-XylNC(CH3)O]4Cl2 towards substitution by N3-, NCS-,
NCO-, H2O, py and 4,4'-bpy has been examined and the crystal structures of the azide and thio-
Rhenium Compounds 297
Walton
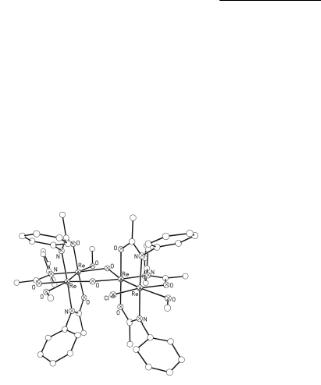
cyanate substituted products determined.169 Studies of the electronic absorption spectra of some of the compounds have shown168 that the βΑβ* transitions are at higher energies than those of Re2(O2CR)4Cl2 molecules. Amidato-bridged compounds that contain the cis-ReN2O2 geometry are prone to react with oxygen in aqueous media; in the case of Re2[µ-PhNC(CH3)O]4Cl2 the unsymmetrical tetranuclear complex {Re4[µ-PhNC(CH3)O]6Cl(µ-O)(µ-OH)(MeOH)3}(ReO4)2 has been isolated when methanol is present.170 This complex contains two quadruply bonded {Re2[µ-PhNC(CH3)O]3}3+ core units (see Fig. 8.11). The Re–Re bond distances in this compound are 2.213(2) Å and 2.200(2) Å; data for this compound are not listed in Table 8.1. Mass spectrometry studies have shown170 that the basic structural integrity of this tetranuclear Re compound is retained in the gas phase.
Fig. 8.11. The structure of the {Re4[µ-PhNC(CH3)O]6Cl(µ-O)(µ-OH)(MeOH)3}2+ cation. The two quadruply bonded Re2[µ-PhNC(CH3)O]3 units are bridged by an oxo and hydroxo ligand; one Cl and three methanol ligands occupy the four remaining coordination sites about the two dirhenium cores.
Dirhenium(III) amidate complexes have also been prepared from (Bu4N)2Re2Cl8 by the hydrolysis of acetonitrile and benzonitrile. The salts (Bu4N){Re2[µ-HNC(CH3)O]2Cl5},171 (Bu4N)2[{Re2[µ-HNC(CH3)O]Cl6}2]172 and (Bu4N){Re2[µ-HNC(Ph)O]Cl6}173 have been structurally characterized (see Table 8.1). In the bis-acetamidate complex the amidate ligands are cis to one another in a head-to-head fashion (i.e. cis-ReN2 and cis-ReO2), although NMR spectroscopy has been interpreted in terms of the structure being best represented as (Bu4N){Re2[µ-HNC(CH3)O][µ-NC(CH3)OH]Cl5} (at least in solution).171 The monoacetamidate complex is linked into a dimer-of-dimers by bridging chlorides.172 The complexes (Bu4N){Re2[µ-HNC(CH3)O]2Cl5} and (Bu4N){Re2[µ-HNC(Ph)O]Cl6} react with Ph2PCH2PPh2 to form the paramagnetic Re25+ compounds Re2[µ-HNC(R)O]Cl4(µ-dppm)2, where R = CH3 or Ph.171,173 Another instance where hydrolysis leads to an µ-amidate complex has been encountered in the reaction of (Bu4N)2Re2Cl8 with 1,4-dicyanobenzene in aqueous ethanol.174 Extraction of the product into DMF enabled crystals of the centrosymmetric diamidate-bridged complex (Bu4N)2{[Re2Cl6(DMF)]2[µ-HNC(O)C6H4C(O)NH]} to be isolated and structurally characterized.
A substitution reaction of a different type involving the dirhenium(III) carboxylates is encountered in the case of the reaction between Re2(O2CCH3)4Cl2 and (Bu4N)MoS4 in acetonitrile.175 This is said to afford (Bu4N)2Re2Mo4S16, in which the Re–Re quadruple bond is believed to be preserved and [MoS4]2- ligands either bridge the dirhenium unit or chelate the individual rhenium atoms.

298Multiple Bonds Between Metal Atoms Chapter 8
8.4.4 Neutral ligands
Although reactions with neutral donors sometimes involve metal-metal bond cleavage as a dominant reaction course, there are many examples where such reactions give products that bear a close structural relationship to [Re2X8]2-. The best known are the phosphine complexes of the type Re2X6(PR3)2, many of which are formed upon reacting methanol solutions of [Re2Cl8]2- and [Re2Br8]2- with monodentate tertiary phosphines (e.g., the series PPh3, PEtPh2, PEt2Ph and PEt3) under mild reaction conditions.22,176-178 The PMe3 complex Re2Cl6(PMe3)2 cannot be obtained by this method, but only by resorting to the one-electron oxidation of the crystalline complex 1,2,7-Re2Cl5(PMe3)3·Bu4NCl,179 which is in turn prepared from (Bu4N)2Re2Cl8 (see
Section 8.5.4).
Re |
Cl |
(PMe |
) |
·(Bu |
NCl) + NOBF |
4 |
CH2Cl2 |
Re Cl |
(PMe |
) |
2 |
+ PMe |
3 |
+ NO + (Bu N)BF |
4 |
|
|
||||||||||||||||
2 |
5 |
3 |
3 |
4 |
|
|
2 |
6 |
3 |
|
|
4 |
||||
A similar procedure has also been used to prepare Re2Cl6(PMe2Ph)2; in this case the NOBF4 oxidation of 1,2,7-Re2Cl5(PMe2Ph)3 is carried out in the presence of one equivalent of added Bu4NCl.180 As we shall see in Sect 8.5.4, it is very easy to obtain reduced Re25+ and Re24+ species by the reaction of (Bu4N)2Re2X8 with monodentate tertiary phosphines, so much so that reduced complexes are often the predominant products. In the case of the reaction between (Bu4N)2Re2Cl8 and PMe3, the only rhenium(III) compound that has been isolated is the edgeshared bioctahedral complex Re2(µ-Cl)2Cl4(PMe3)4, in which there is no metal-metal bond (the Re–Re distance is 3.8476(4) Å).179 Bioctahedral dirhenium(III) complexes have also been isolated with other phosphines, and in some instances they may be intermediates in reactions where lower oxidation state complexes are formed by a disproportionation process; examples of these bioctahedral species include (Bu4N)2[Re2(µ-PPh2)2Cl6(PPh2H)2],181 (Bu4N)[Re2(µ- Cl)2Cl5(PEt3)3],182 Re2(µ-PEt2)2Cl4(PEt2H)4183 and Re2(µ-I)2I4(PMe3)4.184 As yet, there is no instance where an iodide complex of the type Re2I6(PR3)2 has been isolated.
Several crystal structure determinations have been carried out on chloro complexes of the type Re2Cl6(PR3)2. On the basis of the ligand atom numbering scheme shown in 8.12, the structures are all of the type 1,7-Re2Cl6(PR3)2 (8.13) and the Re–Re distances span the narrow range 2.208-2.227(1) Å.179,180,185-188
8.12 |
8.13 |
An interesting structural variation is encountered in the case of the mixed halide-alkox- ide complexes Re2X4(OR)2(PPh3)2 and Re2Cl3(OEt)3(PPh3)2.98,141-143 As mentioned already in Section 8.4.2 these compounds are, in reality, the mixed-valent dirhenium(IV,II) species (RO)2X2ReReX2(PAr3)2 in which a Re–Re quadruple bond is still preserved (see Table 8.1 for structural information). Formally, they are derivatives of the 1,3-Re2Cl6(PR3)2 isomers that have not yet been isolated with monodentate phosphines. Indeed it has been pointed out that the syntheses of 1,2- and 1,3- isomers of Re2Cl6(PR3)2 are improbable except by resorting to chelating diphosphines.189 This has been accomplished in the case of the reaction of (Bu4N)2Re2Cl6 with the chelating phosphine 1,1'-bis(diphenylphosphino)ferrocene.190 The

Rhenium Compounds 299
Walton
structure of Re2Cl6(dppf) is shown in Fig. 8.12 and is clearly an example of a 1,3-Re2Cl6(PR3)2 molecule. What is almost certainly a very close structural analog of Re2Cl6(dppf) is isolated by the reaction of (Bu4N)2Re2Cl8 with bis[2-(diphenylphosphino)phenyl]ether.191 Its spectroscopic and electrochemical properties are very similar to those of Re2Cl6(dppf); it only differs from Re2Cl6(dppf) in having a weak axially bound ether O atom in place of the unbound Fe atom of Re2Cl6(dppf).191 The salt (Bu4N)Re2Cl7(P O P) is formed when 4,6-bis(diphenylphosphino) dibenzofuran is reacted with (Bu4N)2Re2Cl8; in this case, the potentially tridentate phosphinoether ligand is probably δ1-phosphine bound, so the compound is structurally similar to species of the type [Re2Cl7(PR3)]- (vide infra).191(b)
Fig. 8.12. The structure of Re2Cl6(dppf).
The kinetics of the stepwise replacement of two chlorides of [Re2Cl8]2- by tertiary phosphine and arsine ligands has been examined.192 Measurements were carried out in dichloromethane solution and involved the ligands PEt2Ph, AsEt2Ph, PBun3-xPhx and AsBun3-xPhx (x = 1-3); the reactions proceed as shown in the scheme below, with k2 > k1 and k-2 >> k-1. All the reactions studied followed second-order kinetics, in accord with associative mechanisms. When these Group 5 ligands are used in large excess, then reduction of the Re26+ core occurs (Section 8.5.4). Through the reactions of Re2Cl6(PR3)2 (PR3 = PBun3, PBun2Ph, PBunPh2 or PPhBzMe) with Ph4AsCl in CH2Cl2, samples of (Ph4As)Re2Cl7(PR3) have been isolated.192,193 The identities of all these salts have been confirmed by X-ray crystallography (Table 8.1).193-195 Similarly, it was found that the reaction of Re2Cl6(AsBun2Ph)2 with Bu4NBr in CH2Cl2 affords (Bu4N)2Re2Cl6Br2.192 The interesting mixed-salt (Bu4N)4[Re2Cl7(PMe3)]2[Re2Cl8], that contains both [Re2Cl7(PMe3)]- and [Re2Cl8]2- anions, has been prepared and structurally characterized.185 It is formed when the Re25+ complex 1,2,7-Re2Cl5(PMe3)3·Bu4NCl is oxidized with NOBF4 in the presence of an additional equivalent of Bu4NCl.185 Examples of the dicationic tetrakis(phosphine) species [Re2Cl4(PR3)4]2+ are also known; these are formed by the two-elec- tron oxidation of Re2Cl4(PR3)4 and are discussed in Section 8.5.4.
In a study of the chemistry of trirhenium(III) cluster alkyls, Wilkinson and co-workers196 discovered that they undergo cleavage reactions to yield dirhenium(III) or dirhenium(II) com-

300Multiple Bonds Between Metal Atoms Chapter 8
plexes. When the methyl derivatives Re3(CH3)9 or Re3(CH3)9(PR3)3 are treated with a large excess of tertiary phosphine, the centrosymmetric quadruply-bonded dirhenium(III) complexes Re2(CH3)6(PR3)2 (PR3 = PMe3, PMe2Ph or PEt2Ph) are produced.196
Anionic dirhenium(III) species have also been obtained with diphosphines. The best characterized example is (Bu4N)Re2Cl7(bdppp) which has an unsymmetrical structure [PCl4ReReCl3N], wherein which an uncoordinate phosphorus atom of the 2,6-bis(diphenylphosphino)pyridine ligand blocks, but does not bind to, the axial position of the coordinatively unsaturated metal center.197 The bidentate ligands Ph2PC>CPPh2 and trans-Ph2PCH=CHPPh2 (abbreviated LL), which are capable of forming intermolecular bridges, react with (Bu4N)2Re2Cl8 in methanol-conc HCl mixtures to give (Bu4N)2[(Re2Cl7)2(µ-LL)], in which pairs of monoanionic [Re2Cl7L]- units are linked through LL bridges.198 Interestingly, when the chelating form of Ph2PCH=CHPPh2 (cis-dppee) is used in place of trans-Ph2PCH=CHPPh2, cleavage of the Re–Re quadruple bond predominates to give trans-[ReCl2(dppee)2]Cl.199 The latter reaction course is commonly encountered when bidentate phosphine and arsine ligands are used that have two bridgehead carbons between the group 5 donor atoms. The best known example is the reaction of (Bu4N)2Re2Cl8 with Ph2PCH2CH2PPh2 in acetonitrile which gives the paramagnetic complex (dppe)Cl2Re(µ-Cl)2ReCl2(dppe).200
A neutral complex that contains only a single bridging ligand is `-Re2Cl6(S,S-isodiop),
where isodiop is the zwitterionic ligand Ph2PCH2CH(O)CHOC(Me)2P(Ph)2CH2.201 It is formed by the reaction of Re2(O2CCH3)2Cl4 with S,S-diop and Me3SiCl in THF, and involves the rearrangement of S,S-diop (S,S-diop is (+)-2,3-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylp hosphino)butane) to S,S-isodiop, a ligand that coordinates through a P and O atom.201 This was the first example of a structurally characterized chiral dirhenium(III) complex.
Another set of reactions that should be mentioned are those between (Bu4N)2Re2X8 and diphosphines in which only a single bridgehead atom separates the phosphorus atoms. This is exemplified by the case of the dark purple, diamagnetic complex Re2Cl6(µ-dppm)2, which is the product of the reaction between (Bu4N)2Re2Cl8 and Ph2PCH2PPh2 in acetonitrile, acetone or dichloromethane.202,203 When an alcohol is used as the reaction solvent, the mixed chloroalkoxides Re2Cl5(OR)(µ-dppm)2 (R = CH3, C2H5, n-C3H7 or n-C4H9) are produced.203 In a theoretical analysis of d4–d4 M2L10 complexes by Hoffmann and his coworkers204 the extreme cases of diamagnetic, unbridged, [Re2Cl8L2]2- type ‘Cotton structures’ with short Re–Re distances, and the paramagnetic di-µ-chloro bridged ‘Walton complexes’, e.g. Re2Cl6(dppe)2 (see Section 8.7), with long Re–Re separations were considered. It was suggested that Re2Cl6(dppm)2, which had first been reported in 1976, might represent the case of an intermediate di-µ-chloro bridged, metal-metal double-bonded structure (μ2/2β*2β2 configuration). A few years later,203 this was confirmed to be the case when the crystal structure of this compound showed it to be Re2(µ-Cl)2Cl4(µ-dppm)2 (8.14). The Re–Re distance of 2.616(1) Å is fully in accord with a Re–Re double bond.203 A crystal structure determination on the ethoxide derivative Re2Cl5(OEt)(µ-dppm)2 showed it to be similar to that of Re2Cl6(µ-dppm)2, with an ethoxide group in place of one of the terminal chloride ligands.203 Subsequently, the isostructural complex Re2Cl6(µ-dmpm)2 was prepared by reacting a CH2Cl2 solution of (Bu4N)2Re2Cl8 with one of Me2PCH2PMe2 in acetone at room temperature; the Re–Re distance is 2.5807(4) Å.205
An interesting property of Re2Cl6(µ-dppm)2 is its rich redox chemistry. The cyclic voltammograms of its solutions in Bu4NPF6-CH2Cl2 show four metal-based couples in the potential range +1.8 to -1.8 V (vs. Ag/AgCl).203,206 Two of these correspond to one-electron oxidations, and two are one-electron reductions. An oxidation at +0.81 V and reduction at -0.54 V can be accessed with the use of NOX (X = BF4- or PF6-) as oxidant and (δ5-C5H5)2Co as reductant to give [Re2Cl6(µ-dppm)2]X and [(δ5-C5H5)2Co][Re2Cl6(µ-dppm)2], respectively.206 The crys-