

Multiple Bonds Between Metal Atoms / 06-X3M _ MX3 Compounds of Molybdenum and Tungsten
.pdf6
X3MɓMX3 Compounds of
Molybdenum and Tungsten
Malcolm H. Chisholm and Carl B. Hollandsworth,
The Ohio State University
6.1Introduction
After the dimetal unit within the cubic X4MMX4 structural motif, the X3M>MX3 compounds of Mo and W provide the most pertinent examples of coordination complexes having metal–metal multiple bonds. For the most part X3M>MX3 compounds have staggered conformations with MMX bond angles within the range 100-105° which has led to the common description as “ethane-like” dimers. This terminology, though descriptively useful, is not totally accurate. There is, to date, no evidence for the existence of their monomeric counterparts, although Cummins and coworkers have made monomeric Mo complexes with extremely bulky amide ligands.1-3
Moreover, unlike substituted ethane derivatives, a number of eclipsed X3M>MX3 ground state geometries are known which calls into question the optimum enthalpic geometry of the X3M>MX3 species. It is, however, a very striking testimony to the strength of MM multiple bonding that X3M>MX3 species exist in preference to the bridged-ligand structures (e.g., X2M2(µ-X)2M2X2) which have been known for many M(III) compounds with attendant uninegative ligands such as amides, alkoxides, halides, and thiolates.4
As a result of the three M–X μ-bonds formed at each metal and the formation of the MM triple bond, each metal attains a share of 12 electrons. With ligands X that are capable of /-do- nation (e.g., amide, alkoxide, or thiolate), the metal atoms may increase their effective electron count and thereby formally satisfy the EAN rule. However, such /-buffering leaves the metal atoms susceptible to nucleophilic attack by μ-donating ligands and the X3M>MX3 compounds commonly increase their coordination numbers through association of neutral Lewis bases. This increase in coordination number can also be achieved by transforming the X- ligands into bidentate ligands such as in the replacement of alkoxides with carboxylates. The inherent coordinative unsaturation of X3M>MX3 compounds allows uptake of a wide variety of substrates by the dinuclear center.
If this substrate is redox active, very interesting and often unusual reactions can be observed. Research in the Chisholm group over the last three decades has elucidated much of the coordination chemistries of X3M>MX3 compounds. This work has also focused on connecting the chemistries of M>M bonds with those of MM single, double and quadruple bonds for M = Mo and W. Singlyand doubly-bonded compounds can be accessed by oxidative-addition reactions
203

204Multiple Bonds Between Metal Atoms Chapter 6
at the M>M bond and, conversely, reductive elimination leads to quadruply bonded species. Much of this closely related chemistry involving the reactivities of X3M>MX3 compounds is summarized within this chapter.
6.2Homoleptic X3MɓMX3 Compounds
6.2.1 Synthesis and characterization of homoleptic M2X6 compounds
The first member in the X3M>MX3 series was Mo2(CH2SiMe3)6 (Fig. 6.1) formed in a metathetic reaction involving Me3SiCH2MgBr and a molybdenum trihalide.5 Also claimed, though without full structural characterization were Mo2(CH2Ph)65, W2(CH2CMe3)66, and W2(CH2SiMe3)6.5 These compounds were formed in metathetic reactions involving MoCl5 or WCl6. The yellow Mo2(CH2SiMe3)6 (m.p. 99 °C) and orange-brown W2(CH2SiMe3)6 (m.p. 110 °C) were volatile and sublimed in vacuo at 10-4 torr at 100–120 °C. These alkyl compounds are hydrocarbon soluble, diamagnetic, and stable to dry air in the solid-state for short periods of time but are oxidized by air in solution. Their NMR spectra indicated only one type of alkyl ligand.
Fig. 6.1. The structure of Mo2(CH2SiMe3)6.
As is now well known, metathetic reactions involving Mo and W halides are extremely complex and lead to the formation of several different compounds via redox reactions and C−H bond activation processes. Subsequently, compounds such as the paramagnetic d1-Mo(V) alkylidene, (Me3SiCH2)3Mo=CHSiMe3 and the diamagnetic alkylidyne bridged complex [(Me3SiCH2)2W]2(µ-CSiMe3)2 were also discovered as products in these reactions.7 Alkyl for alkoxide group exchange involving M2(OR)6 compounds proved a cleaner route to the homoleptic M2R6 compounds. This was first noted by Rothwell8 and further explored by Gilbert.9 Bonding parameters for the structurally characterized homoleptic alkyls are given in Table 6.1. Notably absent are homoleptic compounds with `-hydrogen-containing alkyl ligands.
Table 6.1. Structural parameters for homoleptic M2X6 compounds |
|
|
|
|||
M |
X |
M–Ma |
M–Xb |
M–M–Xc |
symd |
ref. |
Mo CH2SiMe3 |
2.167 |
2.13 |
100 |
s |
5 |
|
Mo CH2SiMe2Ph |
2.170 |
2.11 |
100 |
s |
10 |
|
Mo |
CH2Ph |
2.175 |
2.16 |
98 |
s |
8 |
Mo CH2CMe2Ph |
2.176 |
2.13 |
98 |
s |
10 |
|
Mo |
(OCHMe2)3N |
2.177 |
1.95 |
99 |
s |
11 |
Mo |
½ NPri(CH2)2PriN |
2.188 |
1.97 |
101 |
e |
12 |
Mo |
½ NMe(CH2)2MeN |
2.190 |
1.97 |
102 |
e |
15 |

X3MɓMX3 Compounds of Molybdenum and Tungsten 205
Chisholm and Hollandsworth
M |
X |
M–Ma |
M–Xb |
M–M–Xc |
symd |
ref. |
Mo |
½ OCMe2CMe2O |
2.194 |
1.89 |
99 |
e |
13 |
Mo |
NMe2 |
2.214 |
1.98 |
103 |
s |
14,15 |
Mo |
Si7O12Cy7 |
2.215 |
1.90 |
102 |
s |
16 |
Mo |
½ (S)-(-)-OCPh2CH2CH2O |
2.217 |
1.90 |
100 |
e |
17 |
Mo |
glucofuranosideg |
2.218 |
1.90 |
102 |
s |
18 |
Mo |
SeC6H2Me3 |
2.218 |
2.44 |
97 |
s |
19 |
Mo |
OCH2But |
2.222 |
1.87 |
103 |
s |
20 |
Mo |
SMes |
2.228 |
2.33 |
97 |
s |
21,22 |
Mo |
OCMe(CF3)2 |
2.230 |
1.88 |
98 |
s |
23 |
Mo |
OCMe2Ph |
2.238 |
1.89 |
101 |
s |
9 |
Mo |
O2Si(OBut)2 |
2.240 |
1.91e |
96 |
e |
24 |
W |
CH2Ph |
2.249 |
2.16 |
98 |
s |
10 |
W |
CH2SiMe3 |
2.255 |
2.14 |
102 |
s |
25 |
Mo |
O-1,4-dipentyl-[2,2,2]-bicyclooctyl |
2.258 |
1.89 |
104 |
s |
26 |
W |
CH2SiMe2Ph |
2.259 |
2.11 |
102 |
s |
10 |
W |
½ NMe(CH2)2MeN |
2.265 |
1.97 |
102 |
e |
13 |
W |
½ OCMe2CMe2O |
2.274 |
1.90 |
100 |
e |
27 |
W |
O2CBut |
2.292 |
2.10e |
96 |
e |
28 |
W |
NMe2 |
2.292 |
1.97 |
103 |
s |
29,30 |
W |
SeMes |
2.300 |
2.43 |
97 |
s |
19 |
Mo |
½ COT |
2.302 |
naf |
naf |
e |
31 |
W |
OCMe(CF3)2 |
2.309 |
1.88 |
101 |
s |
9 |
W |
SMes |
2.312 |
2.32 |
97 |
s |
22 |
W |
OPri |
2.315 |
1.87 |
106 |
s |
32 |
W |
OSiMe2But |
2.324 |
1.93 |
100 |
s |
33 |
W |
OBut |
2.333 |
1.89 |
109 |
s |
h |
W |
glucofuranosideg |
2.334 |
1.87 |
109 |
s |
18 |
W |
(+)-mentholate |
2.338 |
1.88 |
104 |
s |
34 |
W |
OCy |
2.340 |
1.87 |
107 |
s |
27 |
W |
O-1,4-dipentyl-[2,2,2]-bicyclooctyl |
2.341 |
1.88 |
105 |
s |
26 |
W |
½ OCMe2CH2CMe2O |
2.360 |
1.87 |
110 |
e |
35- |
|
|
|
|
|
|
37 |
W |
½ 1,4-TMS2-COT |
2.363 |
naf |
naf |
e |
h |
W |
½ COT |
2.375 |
naf |
naf |
e |
31 |
a |
Å, ± 0.001 Å |
b |
Å, ± 0.01 Å |
c |
°, ± 0.1 ° |
d |
s = staggered, e = eclipsed |
e |
disregarding axial coordination |
f |
for COT compounds there are a variety of M–C bond lengths ranging from 2.22(1) to 2.56(1) Å |
g |
anion of 1,2:5,6-di-O-isopropylidene-_-D-glucofuranose |
h |
M. H. Chisholm, J. C. Gallucci, and C. B. Hollandsworth, unpublished results |
The amides M2(NMe2)6 were subsequently discovered14, 15, 38-41 and based on the well-known alcoholysis of metal amides, the alkoxides M2(OR)6 were synthesized.20, 42-44 The original preparation of the dimetal amides involved reactions of metal chlorides MoCl3, MoCl5, WCl4, or WCl6 with LiNMe2 in mixed hydrocarbon/ether solutions.15,30 Again, the reactions were complex, giving many products such as mononuclear W(NMe2)645 and Mo(NMe2)4.46,47 The compounds Mo(NMe2)4 and Mo2(NMe2)6 were separable by their different volatilities. Mo(NMe2)4

206Multiple Bonds Between Metal Atoms Chapter 6
sublimes at c. 40–60 °C at 10-2 torr whereas Mo2(NMe2)6 sublimes at c. 80–100 °C. W2(NMe2)6 has similar volatility to W(NMe2)6 and both sublime at c. 80–100 °C at 10-2 torr. The pure mononuclear amide is rather sparingly soluble in hydrocarbon solvents, giving ruby-like cubic crystals45 but mixtures of the mononuclear and dinuclear amides co-crystallize giving crystals of W2(NMe2)6·W(NMe2)6.30 Crystals of pure W2(NMe2)6 have also been obtained.30 Because these compounds crystallize in different space groups, a rapid unit cell determination can differentiate between the three species once they are separated. Also, W2(NMe2)6 is pale yellow whereas W(NMe2)6 is red and the 1:1 crystals appear orange.
Improved syntheses of the dinuclear amides followed by using different halide starting materials, MoCl3(dme)23 and NaW2Cl7(thf)5.6 The pure M2(NMe2)6 compounds when sublimed are fluffy and pale yellow powders. They are soluble and stable in dry, deoxygenated hydrocarbon solvents but extremely reactive to air in both the solid-state and in solution. Attempts to prepare other dinuclear, homoleptic amides usually failed except in the case of Mo2(NMeEt)6 and Mo2(NEt2)6 which are well-characterized.15 With the more bulky NEt2 ligand the mixed chloro/amido compound W2Cl2(NEt2)4 was also well-characterized.48 Subsequently, others working with bulky primary amides have obtained similar dinuclear amido-chlorides having MM triple bonds.49-51
The molecular structure of Mo2(NMe2)6 is shown in Fig. 1.7 (see page 16). The molecule has virtual D3d symmetry and the NMe2 ligands are arranged so that six Me–N bonds lie over the M>M bond, and six lie away. These proximal and distal methyl groups exhibit markedly different chemical shifts in their 1H and 13C NMR spectra because of the large magnetic anisotropy induced by the M>M unit. Those lying over the triple bond are deshielded (β 4 ppm in 1H NMR) and those lying away are shielded (β 2 ppm) relative to the typical chemical shift (3 ppm) for a metal amide.52 However, rapid rotation about the M–N bond occurs on the NMR time scale at high temperatures (c. 70 to 80 °C), giving a single resonance as an average of the proximal and distal chemical shifts. From dynamic NMR line broadening and coalescence behavior the activation energy for rotation about the M–N bond has been estimated to be 12 kcal mol-1 for Mo2(NMe2)6. The 1H NMR spectrum of Mo2(NMeEt)6 shows similar but slightly more complex variable temperature behavior as a result of the interconversion of several rotamers having different ratios of proximal and distal methyl and ethyl groups.15
Reactions employing WCl4(Et2O)2 or MoCl3(dme) and the lithium salt of N,N -dimeth- ylethylenediamine led to the isolation of the pale yellow, crystalline M2(MeNCH2CH2NMe)3 compounds in which the bidentate diamide spans the M>M bond and leads to a near eclipsed M2N6 skeleton.13 The eclipsed geometry arises from the strain formed within the resulting six-membered ring incorporating the M>M bond. A related N,N -diisopropyldiamide, Mo2(PriNCH2CH2NPri)3, was recently reported as the product from the reaction between MoCl3(dme) and the dilithioamide salt. It too had an eclipsed structure.53 The initial preparation of these near-eclipsed M2N6-containing dimers was prompted by the prediction of Albright and Hoffmann that M2X6 compounds should be eclipsed in order to maximize the MM bonding.54 As seen in Table 6.1 the MM bond distances are slightly shorter for these compounds.
Homoleptic, dinuclear alkoxides, M2(OR)6, can be obtained by the addition of at least six equivalents of alcohol to the M2(NR2)6 amides:20
M2(NMe2)6 + 6ROH Α M2(OR)6 + 6HNMe2
In several instances, competing reactions occur and only tetranuclear alkoxide clusters are obtained by this method. These clusters will be discussed in Section 6.5. However, the use of bulky, usually tertiary alkoxides guarantees the formation of dinuclear species. In the case of molybdenum, a fairly extensive series of Mo2(OR)6 compounds was isolated via this route,

X3MɓMX3 Compounds of Molybdenum and Tungsten 207
Chisholm and Hollandsworth
namely for R = CH2But, Pri, But, CHMePh, SiMe3, and SiEt3.20 Subsequently, Gilbert noted that several fluorinated alkoxides could be prepared by the direct reaction between MoCl3(dme) and the lithium or sodium alkoxide, thus avoiding the metal amide intermediate altogether.23 Less sterically demanding alcohols such as methanol, ethanol, or n-propanol react with M2(NMe2)6 to generate Mo4(OR)16 compounds in which the M>M bond is no longer present.
The alcoholysis reactions of W2(NMe2)6 are more complex for several reasons:
1.The W>W bond is more labile toward oxidation than the Mo>Mo bond.
2.The W>W bond is more labile toward dimerization to form W4 clusters.
3.W2(OR)6 complexes are more Lewis acidic than their molybdenum counterparts, and thus they more easily form adducts with Lewis bases.
4.The W alkoxides are thermally unstable above c. 60 °C.
This having been stated, a number of W2(OR)6 compounds are now known, the most useful being W2(OBut)6 which despite forming dark red, needle-like crystals have only recently been properly characterized on crystals grown from a thf/ButOH mixture.56 The asymmetric unit of this polymorph of W2(OBut)6 contains 1.5 molecules. The dimer that is contained within the unit cell has one main W–W orientation that comprises 80% of the W2 electron density and four other orientations of approximately 5% each. The half-dimer has one, almost 100%, W–W orientation and this dimer provides the most reliable bonding parameters for
W2(OBut)6.
Closely related compounds, such as W2(O-c-C6H11)6 and W2(OSiMe2But)6 have been structurally characterized and those along with other structurally characterized W2(OR)6 compounds are listed in Table 6.1. Notable among the later synthesized alkoxides of (W>W)6+ are those with chiral groups, such as (+)-D-menthol, that may allow the ditungsten template to act as a chiral Lewis acid, as was also noted by Heppert, et al. in their synthesis of mixed alkoxide/binolates.57 Whereas most Mo2(OR)6 alkoxides are volatile and sublime at c. 60–100 °C and 10-2 torr, the W2(OR)6 alkoxides tend to decompose under such conditions, so the preferred purification method for the alkoxides of tungsten is crystallization from hydrocarbon solvents.
The complexities of these alcoholyses are exemplified by the reaction between W2(NMe2)6 and PriOH. This reaction has been shown to give W2(OPri)6(HNMe2)2,58 the carbide W4(C)(NMe)(OPri)12,59 and the bis-hydride W4(H)2(OPri)1255,60 along with homoleptic alkoxides W2(OPri)6 and W4(OPri)12.32 A better route to (W>W)6+ compounds having secondary and primary alkoxides involves the alcoholysis of W2(OBut)6 whereby ButOH is liberated and most easily removed from the product mixture under reduced pressure as a hydrocarbon azeotrope. This method was used in the low temperature preparation of W2(OPri)6 where W4(OPri)12 cluster formation is kinetically retarded.61 At higher temperatures, however, this reaction gives the tetranuclear clusters or their alcohol adducts, W4(OR)12(HOR).62
Reactions involving pinacol, Me2C(OH)C(OH)Me2 gave the yellow pinacolate complexes M2(OCMe2CMe2O)3, which like the ethylenediamides, have structures in which the central M2O6 skeleton is nearly eclipsed.27 The majority of other structurally characterized M2(OR)6 compounds have staggered M2O6 skeleta except when factors associated with the packing of extremely bulky ligands give a nearly eclipsed skeleton as noted in Table 6.1.
Also included in Table 6.1 are data for the complexes formed in reactions with triols11 and trisilylanols.16 These reactions result in M2L2 complexes where the tridentate ligand L chelates to one metal and spans the MM bond to occupy one coordination site of the other metal as depicted in 6.1. The carbohydrate derivatives prepared by Floriani are closely related to these triolate structures.18
The homoleptic compounds W2(O2CBut)6 and M2COT3 (where M = Mo, W) are also listed in Table 6.1. The carboxylates act as bidentate ligands and in these compounds, each metal

208Multiple Bonds Between Metal Atoms Chapter 6
atom forms five M–O bonds in the plane perpendicular to the MM axis together with an additional, weak, axial W–O bond. There are two O2CR ligands spanning the MM bond.
6.1
In the M2COT3 complexes, one COT ligand straddles the MM bond such that five carbon atoms are within bonding distance of each metal. The other two COT ligands are termi- nally-bound in an δ4-fashion resembling a butadiene ligand, as seen for the recently synthesized 1,4-bis-trimethylsilyl-substituted compound shown in Fig. 6.2.63 The MM bond order in M2COT3 has been variously described as quadruple or double based on the diamagnetism of the compound and qualitative electronic structure arguments. However, recent DFT calculations suggest that this may be viewed as a M>M triple bond and, as can be seen from Table 6.1, the MM distances are closer to those of the M2X6 species than those of MM quadruple or double bonds.64
Fig. 6.2. The structure of Mo2(COT-TMS2)3.
The homoleptic mesityl thiolates and selenates, M2(EC6H2Me3)6 have been prepared via similar metathesis reactions using the mesityl-thiol21,22 or selenol.19,65 The thiolates (red) and selenates (red-brown) are crystalline and have staggered M2E6 skeletons. Attempts to obtain suitable crystal structures of other thiolates such as M2(SBut)6 have been unsuccessful, possibly due to the same issues of W–W disorder that appear in the structure of W2(OBut)6.
6.2.2 Bonding in M2X6 compounds
As can be seen from an inspection of Table 6.1 the MM bond distances in the homoleptic M2X6 compounds span a small range of about 2.15 to 2.35 Å. For a related pair of compounds, the MM distance is longer by c. 0.08 Å for the W compound despite the fact that the M–X bond distances (where X = C, N, O, S, or Se) are either comparable or slightly shorter. The MM distances are roughly 0.1 Å longer than those seen in MM quadruply bonded compounds.4 The origin of the longer WɓW distance compared to MoɓMo, is almost certainly due to increased core-core repulsions. There have been both experimental66 and theoretical67 attempts to estimate the bond strength of MM triple bonds. Although there are uncertainties that arise with

X3MɓMX3 Compounds of Molybdenum and Tungsten 209
Chisholm and Hollandsworth
each approach, a reasonable numerical value of 60 and 90 kcal mol-1 is accepted for the M>M bond strength, where M = Mo and W, respectively.
The bonding in these M2X6 compounds can be considered qualitatively as follows. Taking the M–M axis as the z-axis, each metal forms three MX μ-bonds using s, px, and py hybrids and forms the MM triple bond using metal dz2 and dxz, dyz orbitals. This leads to the formation of a cylindrical triple bond of MM configuration μ2/4 for these d3-d3 complexes. In addition, each metal may use its dxy and dx2-y2 orbitals to form /-bonds to the X ligands (if they are available and of suitable energy). For NR2 ligands, these are oriented in such a way as to maximize amide to metal /-bonding, though only two /-bonds delocalized over three M–N μ-bonds can be formed from ligand to metal /-donation. A consideration of the M–N bond distances (1.96 Å on average) in relation to related M–Csp3 bond distances (2.14 Å on average) certainly supports the importance of this /-donation. In the case of alkoxides the MOC angles are normally in the range 130–150° and the alkyl groups are disposed in either a proximal or distal manner with respect to the M–M bond. In this case, oxygen p/ donation can occur and the relatively short M–O distances (1.88 Å on average) support this view. A similar argument can be made for the thiolates and selenates, but based on M–E distances (E = S, Se) this /-donation is believed to be less important than for the alkoxides. The M2X6 compounds are therefore electronically of the 18-electron count, though the amide and alkoxide ligands provide a /-buffering effect.
Electronic structure calculations have been undertaken on these M2X6 compounds and generally support the preceding qualitative bonding description.68 In C3-symmetry there can be extensive mixing of MX μ and the MM bonding and antibonding orbitals. This is particularly prominent when the metal and ligand orbitals are of similar energy as is the case for the homoleptic alkyls. In the case of alkoxides, the more electronegative oxygen gives a greater energy separation and the photoelectron spectra of M2(OR)6 compounds reveals that the first ionizations can be assigned to ionizations from the MM /- and μ-based orbitals. Ionization from the /-orbitals requires roughly 1 eV more energy than from the β-bond of a MM quadruple bond. In the case of the M2(NR2)6 compounds the ionization from the nonbonding NR2 lone pair / combinations is close in energy to the ionization from the MM /-bonding orbital. Calculations on model thiolates M2(SH)6 also indicate that the HOMO is a sulfur-based lone pair combination. The influence of Xp/ to Md/ donation is to raise the energy of the HOMO, which is the MM /-bonding MO, and it has been suggested that this labilizes the M>M bond in M2(OR)6 compounds.69 The LUMO in these compounds is a metal-based /*β combination which too has some ligand p/ contribution. The color of these compounds, yellow to red, arises from the HOMOΑLUMO transition which may be viewed as a MM / to /* transition.27
Raman spectra have been recorded for some of these M2X6 compounds with the intent of identifying and quantifying the MM stretching frequency. The earliest attempt examined the compounds M2(NMe2)6 and M2(NMe2-d6)6 and concluded that it was not possible to identify a Raman band uniquely associated with ι(MM).15,30 The bands in the region expected for ι(MM) and ι(MN) all showed significant shifts upon deuteration. Subsequent work by Dallinger, Gilbert, and coworkers who examined both M2(CH2EMe3)6 (where E = C or Si) and M2(OR)6 (where R = But, But-d8, CMe2CF3 and 1-adamantyl) were able to assign ι(MoMo) in the range 360–380 cm-1 and ι(WW) from 274–304 cm-1.26 Based on the known values of ι(MM) in qua- druply-bonded complexes their numerical values appear very reasonable and for related pairs of compounds the ratio ι(MoMo)/ι(WW) is found to be 1.30, close to that predicted assuming an equivalent force constant for each triple bond, namely 1.38. This work has subsequently been extended to include computation of the Raman bands and a re-evaluation of the spectra of the M2(NMe2)6 compounds.26

210Multiple Bonds Between Metal Atoms Chapter 6
6.2.3 X3MɓMX3 Compounds as Molecular Precursors to Extended Solids
Some of the M2X6 compounds have been examined as molecular precursors to ceramic materials. Upon heating W2(OBut)6 to 200 °C under a stream of dry N2, the alkoxide decomposes to give WO2 with the elimination of isobutylene, ButOH, and trace amounts of water.70 The Mo2(OBut)6 species is more thermally robust, but in the presence of trace amounts of water decomposes to MoO2 at c. 250 °C. Mo2(OPri)6 sublimed at 120 °C under 1 atm of N2, but W2(OPri)6 decomposed to give a mixture of tungsten metal and W2C.71 A similar product mixture was obtained in the decomposition of W2(O-c-C6H11)6 whereas the Mo analog gave exclusively Mo2C. The introduction of benzyl ligands in the compounds M2(CH2Ph)2(OR)4 triggered decompositions at lower temperatures, around 120 °C, and significantly changed the product distribution.71
In a somewhat related study, Tilley and Su showed that the siloxide Mo2[O2Si(OBut)2]3 decomposed upon heating to give MoO2 as the only crystalline phase, whereas the W analog gave W(s) and WO2.24 However, in both decompositions an amorphous ceramic phase was formed containing metallic Mo or W along with Si and O. McCarley and coworkers have also investigated the use of Mo2(OR)6 compounds and Mo2(NMe2)6 in a sol-gel approach to forming Mo2O3.72 They were unsuccessful in this attempt, but did discover MoO(OH), a new species that could be converted to Mo3O5 and LiMoO2 (along with other molecular species such as Mo3(OH)9(NMe2)·½HNMe2) by reacting with Li2CO3.
6.3M2X2(NMe2)4 and M2X4(NMe2)2 Compounds
A large class of compounds of the general formula 1,2-M2X2(NMe2)4 is now known and the majority has been prepared from metathetic reactions involving 1,2-M2Cl2(NMe2)4. These important starting materials are prepared from the reaction between two equivalents of Me3SiCl and M2(NMe2)6 compounds in a hydrocarbon solvent, typically hexane, benzene or toluene. If a concentrated or near saturated solution of the M2(NMe2)6 compound is employed, the 1,2- M2Cl2(NMe2)4 compounds are formed as orange microcrystalline powders.73 Evidence has been presented that these reactions are catalyzed by adventitious HCl or HNMe2 which allow for the replacement of the NMe2 ligands as seen in the equations below. However, oxidative addition of Me3SiCl followed by reductive elimination of Me3Si–NMe2 is also possible though unlikely.74
Me3SiCl + NHMe2 Me3SiNMe2 + HCl
M2(NMe2)6 + HCl M2Cl(NMe2)5 + HNMe2
If an excess of Me3SiCl is employed, further chloride for amide exchange occurs leading to insoluble products. The M2Cl2(NMe2)4 compounds can be further purified by sublimation. In solution and in the solid-state, the dichlorides adopt the anti rotamer and the barrier to M–N bond rotation is notably higher than that in the M2(NMe2)6 compounds.73 The 1,2- M2Cl2(NMe2)4 compounds have been employed as starting materials in a large number of metathetic reactions employing organolithium or Grignard reagents, leading to the isolation of a wide range of compounds of the type 1,2-M2R2(NMe2)4. Examples include R = Me,41 Et,75 Bu,76 Pri,76 Bui,76, Ph,77 o- and p-tolyl,77 CH2Ph,77 CPh3,78 CH2SiMe3,76 CH2CMe3,76 CH(SiMe3)2,79 SiPh3,78 GePh3,78,80 Si(SiMe3)3,81 SnPh3,78 Sn(SnMe3)3,77,81 PBut2,82 AsBut2,83 OCPh3,84 OSiPh3,84 OB(mesityl)2,85 SBut,86 Cp,87,88 C5H4Me,89 indenyl,87,89 allyl,90 and 3-methylallyl.90 A number of bridging groups have also been employed, e.g., X2 = 1,1 -(C5H4)2Fe,91 (-CH2-)492, and COT.93 Notably absent in this series are alkenyl and alkynyl complexes and attempts to prepare such compounds have always resulted in the formation of insoluble byproducts.94 Reactions employing LiCPhCPhCPhCPhLi and 1,2-W2Cl2(NMe2)4 gave W2(NMe2)4(µ-CPh)(µ-C3Ph3) via C–C

X3MɓMX3 Compounds of Molybdenum and Tungsten 211
Chisholm and Hollandsworth
reductive cleavage and it is likely that some similar reaction occurs in reactions employing alkynyl and alkenyllithium reagents.94
The majority of the 1,2-M2R2(NMe2)4 compounds where R represents a μ-carbon bonded ligand exist as a mixture of anti and gauche rotamers in solution that interconvert slowly on the NMR timescale with ¨G& of 20-24 kcal mol-1. One rotameric form of 1,2-M2R2(NMe2)4 tends to be the most stable due to steric factors. In fact, gauche 1,2-Mo2[CH(SiMe3)2]2(NMe2)4 is air-stable in the crystalline state for days at room temperature, which is a result of the presence of the bulky CH(SiMe3)2 groups.95 The 1,2-ortho- and 1,2-para-tolyl compounds revealed that there is an extremely low barrier to rotation about the M–C μ bond in contrast to the M–N bonds consistent with the view that the latter arises from electronic considerations, Np/ to Md/ donation, and not from steric factors. In fact, the solid-state structure of the ortho-tolyl complex showed that the C6 plane was offset 90° with respect to alignment with the MM axis. Another notable feature of the `-hydrogen-containing alkyl groups is their thermal stability. Many can be sublimed at temperatures near 100 °C at 10-2 torr and they are relatively inert to decomposition by `-H elimination processes despite the fact that the metal-atoms are formally unsaturated. This has been attributed to the important role of the /-donor ligands in bonding to metal d-orbitals that otherwise would be available for (CH)–M interactions.
For allyl, Cp, and indenyl ligands, the focus of attention was on the relative /-donor properties of the ligands. In all cases, δ3-coordination was observed which suggests that Cp and indenyl ligands compete effectively with dimethylamides as /-donors. In the case of allyl ligands, a bridged structure was observed with a relatively long WW distance for a (M>M)6+ compound (2.48 Å). Electronic structure calculations imply that there is a significant interaction between all three allyl /-MOs at the (M>M)6+ center and, in particular, /3 of the allyl can receive electron density from the MM /-orbitals. A similar kind of bonding description can be formulated for W2COT(NMe2)4 where the COT ligand spans the WW triple bond. In solution, the COT ligand is evidently fluxional on the NMR time-scale and rotation occurs by a 1,2-site exchange in a similar manner to that seen recently in the corresponding alkoxides,W2COT(OR)4.96
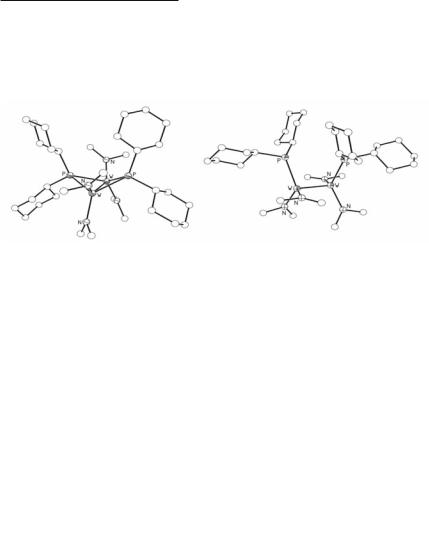
From reactions between W2Cl2(NMe2)4 and two equivalents of LiPR2, the compounds 1,2-W2(PR2)2(NMe2)4 have been isolated and fully characterized.97,98 For R = But, the relatively long W–P bond distances and the pyramidal coordination at phosphorus clearly indicate that Pp/ to Wd/-bonding is less significant than Np/ to Wd/ bonding. In the case of R = Ph, bridged compounds are formed and for R = cyclohexyl, both unbridged and bridged isomers were obtained and shown to interconvert:
1,2-W2(PCy2)2(NMe2)4 W2(µ-PCy2)2(NMe2)4
Similar bridged structures were seen for W2(PPh2)2(NMe2)4 and W2(PPh2)2(OBut)4. The structures of bridged and unbridged molecules are compared in Fig. 6.3. Most notable in the bridged isomer is the non-planar W2P2 unit. The origin of this puckering was traced to electronic factors where d3-d3 MM bonding is maximized.
The compounds M2(NMe2)6 are also labile to reactions with REH where E is a chalcogen and R is an extremely bulky and/or strongly electron withdrawing group. The replacement of NMe2 ligands leads to compounds of the form M2(NMe2)2(ER)4. The series where M = W, E = O and R = CMe2CF3, CMe(CF3)2 and C(CF3)3 was studied in detail to ascertain the influence of the fluorinated alkyl substituents.99 These studies included single crystal X-ray, VT NMR and UV-visible spectroscopy. The introduction of the CF3 groups has a pronounced effect in stabilizing the MM / and μ and Np/-based ionizations and this effect increases with the successive replacement of each methyl by trifluoromethyl. Also in these compounds, the lowest energy ionization bands clearly reveal the removal of the degeneracy of the MM / MO’s.
212Multiple Bonds Between Metal Atoms Chapter 6
As a result of the poor /-donation from the fluorinated alkoxides, the remaining NMe2 ligands /-donate more strongly as evidenced by higher M–N rotational barriers and shorter M–N bond distances. The average M–N bond length is 1.91 Å in W2(NMe2)2(OCMe(CF3)2)4 compared to a W–N bond length of 1.96 Å in W2(NMe2)6.
Fig. 6.3. Structural comparison of bridged and unbridged isomers of W2(PCy2)2(NMe2)4.
6.4Other M2X2Y4, M2X6-n Yn and Related Compounds
Compounds in which the central (M>M)6+ unit is supported by a set of mixed uninegative ligands of the form M2X6-nYn constitute the largest group of compounds having M>M bonds. The ligands X and Y may be monodentate, e.g., as in alkyl, amide and alkoxide, or bidentate as in a carboxylate or `-diketonate and may bind to the binuclear center by spanning the metal–metal bond or by chelating to one metal center. In the latter case, the metal center expands its coordination number first in the equatorial plane and then axially as was seen earlier for W2(O2CNMe2)6108 and W2(O2CBut)628. Most compounds of this type have the formula M2X2Y4 and nearly all are symmetrically substituted about the M2 unit. There are, however, notable exceptions even though the isomerization of 1,1- and 1,2-M2X2Y4 (6.2 and 6.3 respectively) isomers has been shown to have a significant kinetic barrier.101
6.2 |
6.3 |
Perhaps the most notable feature of this class of compounds is the virtual absence of members in which one or more of the groups directly bridge the two metal atoms as is so common for the amide, alkoxide, halide and thiolate ligands. Only for some phosphide groups is µ-PR2 bonding thermodynamically preferred. This again testifies to the energetic importance of preserving the MM μ2/4 electronic configuration in these d3-d3 dinuclear complexes. In some cases, the bonding mode of the ligand X is δn or µ-δn,δn, as for certain hydrocarbon ligands such as Cp, indenyl, allyl or COT. In all of these compounds, the MM bond distances span a very small range from 2.2 to 2.4 Å and for otherwise equivalent complexes, the MoMo distances are shorter than their WW counterparts by roughly 0.08 Å. Table 6.2 summarizes pertinent structural parameters for M2X2Y4 compounds while others such as M2XaYbZc (where a + b + c = 6) are presented in Table 6.3. Table 6.4 summarizes data regarding the M–NR2 rotational barriers in some compounds of the form M2X2(NR2)4 where X is a variety of ligands; R = Me/Et.