

72. Типы химической связи. Гибридизация орбиталей.
Хим. связь- различные виды взаимодействия, обусловливающие устойчивое существование двух многочисленных молекул.
Условием образования химической связи является уменьшение потенциальной энергии системы взаимодействующих атомов.
- Ковалентная. Характеризуется энергией, связь возможна только при выигрыше энергии и направленность. связи, насыщенностью и валентным углом.
Если общеэлектронная пара принадлежит в равной степени обоим атомам, то это связь ковалентно-неполярная, она характерна для молекул газообразных веществ.
Если общеэлектронная пара смещена к более электроотрицательному элементу, то такая связь ковалентно-полярная.
Количественно она определяется дипольным моментом:
ϻ =g
l
=g
l
- Ионная связь – предельный случай ковалентно-полярной связи – характеризуется ненасыщенностью, а огранич. размерами атомов.
NaCl
Такой вид связи «работает» только в объеме кристалла.
-Донорно-акцепторная (ков.связь, ее механизм) – связь, образованная парой электронов принадлежащий одному атому и незаполненная пустой орбиталью.
Этот вид связи характерен для комплексных соединений.
-Металлический
По данному виду связи образованны кристаллы металлов. Это связь между «коллективом» электронов и томами (ионами) металлов
-Межмолекулярные виды связи:
1) ориентационные – взаимодействие между дипольными молекулами, которые при столкновении ориентируются противоположными знаками.
2) дисперсионные – образование хим. связи между молекулами в силу того, что при сталкновении происходит смещение эл. облака первого атома к положительному ядру другого.
3) индукционные – взаимодействие между нейтральными неполярными молекулами и диполями; диполи смещают эл.облако нейтральных, что приводит к электростатическому взаимодействию.
При перекрывании эл.орбиталей разной формы происходит их гибридизация, если орбитали относятся к одному и тому же энергетическому уровню.
Гибридизация – это выравнивание связующих орбиталей по энергии и по форме.
sp-гибридизация.
Явление гибридизации определяет пространственную структуру, образовавшейся молекулы.
37. Окислительно-восстановительные процессы. Взаимодействие Zn и Cu с разбавленной серной кислотой.
Реакции , в результате которых изменяются степени окисленности элементов, называются окислительно-восстановительными.
2Na +nCl2= 2NaCl
Процесс отдачи электронов, сопровождающися повышением степени окисленности элемента называется окислением.
Присоединение электронов, сопровождающееся понижением степени окисленности элемента называется восстановлением.
Вещество, в состав которого входит окисляющий элемент называется восстановителем, а вещество, содержащее восстанавливающий элемент, окислителем.
Число электронов, отдаваемых молекулами (ионами,атомами), восстановителя, равно числу электронов, присоединяемых молекулами (ионами, атомами) окислителя.
Важнейшие восстановители – это металлы (алюминий магний цинк) так же сильными восстановителями являются (С, СО)
Соединения, содержащие элементы в промежуточных степенях окисленности , обладают окислительно-восстановительной двойственностью – способностью вступать в реакции как с окислителями так и с восстановителями.
Так же обладают свойством – самоокислением – самовосстановлением – процесс, в ходе которого часть элемента окисляется, а часть восстанавливается.
Разбавленная и концентрированная серные кислоты ведут себя по-разному.
С Цинком(активный металл может образовывать сернистый газ., элементарную серу и даже сероводород):
2H2SO4 + Zn = SO2 (выделяется) + ZnSO4 + 2H2O
Медь – менее активный металл. При взаимодействии восстанавливает ее до сернистого газа.
2H2SO4 конц + Cu = SO2 (выделяется) + CuSO4 + 2H2O
38. Окислительно-восстановительные реакции. Условия их проведения.
Начало в 37.
Условия проведения ОВР:
43. Основные положения химической кинетики. Скорость химической реакции. Обратимые и необратимые реакции. Принцип Ле Шателье.
Химическая кинетика – раздел химии, который изучает скорость химической реакции и ее механизмы.
Скорость химической реакции показывает изменение концентрации реагирующих веществ в единицу времени, в единицу объема.
V=делта С/ дельта t
Зависимость скорости реакци от конценрации реагирующих веществ
Реакция: А+В=С
V= k [A][B] где к- коэфициент пропорциональности или же константа скорости
Полученное соотношение выражает закон действия масс для химической реакции. Протекающей при сталкновении 2 частиц: при постоянной температуре скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ.
Если 2А+ В = А2В то v= k [A]2[B]
Зависимость скорости реакции от температуры и природы реагирующих веществ:
Вант Гофф
При нагревании на 10 градусов скорость химической реакции увеличивается в 2-4 раза
V2/V1= γt2-t1/10
Это правило действует при незначительных температурах не более 500 градусов
Для более объективной оценки применяют понятие энергоактивности – это та энергия, которой необходима обладать молекула, чтобы при сталкновении их друг с другом произошло взаимодействие
К2/К1= А ҽЕакт/RT где R – универсальная газовая постоянная а Т – температура
Закон Вааге:
Скорость химической реакции пропорциональнаконцентрации реагирующих веществ, взятых в степени их стехеометрических коэфициентов.
аА+вВ=сС + dD
Vпрямая= k пр [A]a[B]b
Vобр= k обр [С]С[D]d
Катализаторы – это вещества ускоряющие процесс химической реакции и не участвующие в ней
Ингибиторы – вещества тормозящие хим.реакцию
Если реакция идет одновременно в разных направлениях, то она называется обратимой.
Если скорость прямой реакции равна скорости обратной наступает динам.равновесие.
Принцип Ле Шателье:
Если на систему, находящейся в динам.равновесии оказывать внешнее воздействие, то она будет смещаться в сторону меньшего этого воздействия.
Для количественной характеристики состояния равновесия введена константа равновесия равная произведению реакци в степени стехиометрического коэфициента на произведение концентраций исходных веществ.
К = Сi продукт/ Ciисход
Сместить равновесие можно если изменить концентрацию веществ, если вещества находятся в газообразном состоянии, то при повышении давления равновесие сместиться в сторону меньшего объема. Если реакция экзотермическая, то повышение температуры.
44 = 72
45. Основы химической термодинамики. Энтальпия и энтропия. Энергия Гиббса.
Химическая термодинамика отвечает на вопросы возможно ли химическое взаимодействие при определенной температуре и давлении.
Термодинамические параметры: р, t, v, m
Термодинамические параметры относятся к стандартным условиям,
Термодинамические функции, которые описываются термодинамическими параметрами
Е – полная энергия системы (потенциальная и кинетическая)
U – внутренняя энергия системы состоящая из энергии всех составных систем.
Существуют системы изолированные (нет обмена массы, Е, информации с окружающейсредой)
Закрытые (возможен обмен одним из параметров – чаще информационный обмен с окружающей стредой)
Открытые (возможен с любым из параметров)
Тепло подведенное к изолированной системе, затрачивается на повышение ее внутренней энергии и совершения работы против внешних сил
Q= дельта U +A
Q- теплота
В процессах, происходящих под постоянным давлением вводится понятие теплосодержания системы Н (энтальпия)
Если дельта Н меньше нуля то процесс возможен
дельта Н = Н конечная – Н начальная
закон Гесса:
тепловой эффект химической реакции дельта Н равен сумме теплового эффекта продуктов минус сумма тепловых эффектов исходных.
Дельта Н = сумма Н прод. – сумма Н исх.
При горении наоборот
Энтропия (S) – мера беспорядка, она зависит от температуры системы, самопроизвольно процесс протекает при ее возрастании.
Дельта S= Q/T
2 закон термодинамики по Больцману
S= k ln W
W- термодинамическая вероятность – число микросостояний, которое достигает макросостояние
S стремится к максимуму
Чем больше энтропия, тем устойчивее система.
Для того, чтоб определить энтропию процесса нужно из суммы энтропий продуктов вычесть сумму энтропий исходных веществ
Дельта S = сумма S прод – сумма S исх
Для удобства определения направления и возможности процесса была введена термодинам. Функция Гиббса – G при р = константа
Свободная энергия – это та часть системы, которая может быть затрачена на выполнение полезной работы.
ТS – диссипация – связанная энергия, которая не может быть затрачена на совершение работы.
Самопроизвольно процесс возможен в изолированной системе если дельта G < 0
46. Основные химические свойства предельных и непредельных углеводородов.
Алка́ны (также насыщенные углеводороды, парафины, алифатические соединения) — ациклические углеводороды линейного или разветвлённого строения, содержащие только простые связи и образующие гомологический ряд с общей формулой CnH2n+2.
Алканы являются насыщенными углеводородами и содержат максимально возможное число атомов водорода.
Алканы имеют низкую химическую активность. Это объясняется тем, что единичные C-H и C-C связи относительно прочны и их сложно разрушить. Поскольку углеродные связи неполярны, а связи С — Н малополярны, оба вида связей малополяризуемы и относятся к σ-виду, их разрыв наиболее вероятен по гомолитическому механизму то есть с образованием радикалов.
Галогенирование
Галогенирование — это одна из реакций замещения. В первую очередь галогенируется наименее гидрированый атом углерода (третичный атом, затем вторичный, первичные атому галогенируются в последнюю очередь). Галогенирование алканов проходит поэтапно — за один этап замещается не более одного атома водорода:
CH4 + Cl2 → CH3Cl + HCl (хлорметан)
Горение
Основным химическим свойством предельных углеводородов, определяющих их использование в качестве топлива, является реакция горения. Пример:
CH4 + 2O2 → CO2 + 2H2O + Q
Разложение
Реакции разложения происходят лишь под влиянием больших температур. Повышение температуры приводит к разрыву углеродной связи и образованию свободныхрадикалов.
Примеры:
CH4 → C + 2H2 (t > 1000 °C)
Крекинг
При нагревании выше 500 °C алканы подвергаются пиролитическому разложению с образованием сложной смеси продуктов, состав и соотношение которых зависят от температуры и времени реакции. При пиролизе происходит расщепление углерод-углеродных связей с образованием алкильных радикалов.
Дегидрирование
Образование:
1)В углеродном скелете 2 (этан) или 3 (пропан) атома углерода — получение (терминальных) алкенов, так как других в данном случае не может получиться; выделение водорода:
Условия протекания: 400—600 °C, катализаторы — Pt, Ni, Al2O3, Cr2O3
а)CH3-CH3 → CH2=CH2 + H2 (этан → этен)
Непредельные углеводороды — углеводороды с открытой цепью, в молекулах которых между атомами углерода имеются двойные или тройные связи. Непредельные углеводороды способны к реакциям присоединения по двойным и тройным связям в открытой цепи. Они, например, присоединяют бром, легко окисляются растворомперманганата калия. Для многих непредельных углеводородов характерны реакции полимеризации. К непредельным углеводородам принадлежит несколько гомологических рядов: этилена (алкены), ацетилена (алкины), диены.
Полимериза́ция — процесс образования высокомолекулярного вещества (полимера) путём многократного присоединения молекул низкомолекулярного вещества (мономера, олигомера) к активным центрам в растущей молекуле полимера.
47. Первый закон термодинамики.
1-й закон — первое начало термодинамики. Представляет собой формулировку обобщённого закона сохранения энергии для термодинамических процессов. В наиболее простой форме его можно записать как δQ = δA + dU, где dU есть полный дифференциал внутренней энергии системы, а δQ и δA есть элементарное количество теплоты, переданное системе, и элементарная работа, совершенная системой соответственно. Нужно учитывать, что δA и δQ нельзя считать дифференциалами в обычном смысле этого понятия, поскольку эти величины существенно зависят от типа процесса, в результате которого состояние системы изменилось.
48. Периодическая система Д.И.Менделеева.
Периоди́ческая систе́ма хими́ческих элеме́нтов (табли́ца Менделе́ева) — классификация химических элементов, устанавливающая зависимость различных свойств элементов от заряда атомного ядра. Система является графическим выражениемпериодического закона, установленного русским химиком Д. И. Менделеевым в 1869 году. Её первоначальный вариант был разработан Д. И. Менделеевым в 1869—1871 годах и устанавливал зависимость свойств элементов от их атомного веса (по-современному от атомной массы). Всего предложено несколько сотен[1] вариантов изображения периодической системы (аналитических кривых, таблиц, геометрических фигур и т. п.).
49.Периодичность изменения свойств химических элементов (s,p,dэлементы)
Зависимость энергии ионизации атома от порядкового номера элемента (рис. 1) носит отчетливо периодический характер. Легче всего удалить электрон из атомов щелочных металлов, включающих по одному валентному электрону, труднее всего — из атомов благородных газов, обладающих замкнутой электронной оболочкой. Поэтому периодичность изменения энергии ионизации атомов характеризуется минимумами, отвечающими щелочным металлам, и максимумами, приходящимися на благородные газы. Наряду с этими резко выраженными минимумами и максимумами на кривой энергии ионизации атомов наблюдаются слабо выраженные минимумы и максимумы, которые по-прежнему нетрудно объяснить с учетом упомянутых эффектов экранирования и проникновения, эффектов межэлектронных взаимодействий и т. д
Наибольшим сродством к электрону обладают p-элементы VII группы. Наименьшее сродство к электрону у атомов с конфигурацией s² (Be, Mg, Zn) и s²p6 (Ne, Ar) или с наполовину заполненными p-орбиталями (N, P, As)
Электроотрицательность
В периодах наблюдается общая тенденция роста электроотрицательности, а в подгруппах — её падение. Наименьшая электроотрицательность у s-элементов I группы, наибольшая — у p-элементов VII группы.
Уменьшение значений орбитальных атомных радиусов при переходе от щелочного металла к соответствующему (ближайшему) благородному газу носит, за исключением ряда Li—Ne, немонотонный характер, особенно при появлении между щелочным металлом и благородным газом семейств переходных элементов (металлов) и лантаноидов или актиноидов. В больших периодах в семействах d- и f-элементов наблюдается менее резкое уменьшение радиусов, так как заполнение орбиталей электронами происходит в пред- предвнешнем слое. В подгруппах элементов радиусы атомов и однотипных ионов в общем увеличиваются.
s- и р-элементы
Так, при переходе от s-элемента I группы к р-элементу VIII группы на кривой энергии ионизации атомов и кривой изменения их радиусов имеются внутренние максимумы и минимумы. Понятно, что экранирование ядра возрастает с увеличением числа внутренних электронных слоев. Поэтому в подгруппах s- и р-элементов наблюдается тенденция к уменьшению энергии ионизации атомов.
В характере изменения свойств s- и р-элементов в подгруппах отчетливо наблюдается вторичная периодичность (рис. 7). Для её объяснения привлекается представление о проникновении электронов к ядру. Как показано на рисунке 9, электрон любой орбитали определенное время находится в области, близкой к ядру. Иными словами, внешние электроны проникают к ядру через слои внутренних электронов. Как видно из рисунка 9, внешний 3s-электрон атома натрия обладает весьма значительной вероятностью находиться вблизи ядра в области внутренних К- и L-электронных слоев.
Концентрация электронной плотности (степень проникновения электронов) при одном и том же главном квантовом числе наибольшая для s-электрона, меньше — для р-электрона, ещё меньше — для d-электрона и т. д. Например, при n = 3 степень проникновения убывает в последовательности 3s>3p>3d , что эффект проникновения увеличивает прочность связи внешних электронов с ядром. Вследствие более глубокого проникновения s-электроны в большей степени экранируют ядро, чем р-электроны, а последние — сильнее, чем d-электроны.
Во внешнем слое у атомов d-элементов (за исключением Pd) находятся 1—2 электрона (ns-состояние). Остальные валентные электроны расположены в (n—1)d-состоянии, т. е. в предвнешнем слое.
Подобное строение электронных оболочек атомов определяет некоторые общие свойства d-элементов[18]. Так, их атомы характеризуются сравнительно невысокими значениями первой энергии ионизации. Как видно на рисунке 1, при этом характер изменения энергии ионизации атомов по периоду в ряду d-элементов более плавный, чем в ряду s- и p-элементов. При переходе от d-элемента III группы к d-элементу II группы значения энергии ионизации изменяются немонотонно. Так, на участке кривой (рис. 1) видны две площадки, соответствующие энергии ионизации атомов, в которых заполняются Зd-орбитали по одному и по два электрона. Заполнение 3d-орбиталей по одному электрону заканчивается у Mn (3d54s2), что отмечается некоторым повышением относительной устойчивости 4s2-конфигурации за счет проникновения 4s2-электронов под экран 3d5-конфигурации. Наибольшее значение энергии ионизации имеет Zn (3d104s2), что находится в соответствии с полным завершением Зd-подслоя и стабилизацией электронной пары за счет проникновения под экран 3d10-конфигурации.
В подгруппах d-элементов значения энергии ионизации атомов в общем увеличиваются. Это можно объяснить эффектом проникновения электронов к ядру. Так, если у d-элементов 4-го периода внешние 4s-электроны проникают под экран 3d-электронов, то у элементов 6-го периода внешние 6s-электроны проникают уже под двойной экран 5d- и 4f-электронов. Например:
22Ti …3d24s2 |
I = 6,82 эВ |
40Zr …3d104s24p64d25s2 |
I = 6,84 эВ |
72Hf… 4d104f145s25p65d26s2 |
I = 7,5 эВ |
Поэтому у d-элементов 6-го периода внешние бs-электроны связаны с ядром более прочно и, следовательно, энергия ионизации атомов больше, чем у d-элементов 4-го периода.
Размеры атомов d-элементов являются промежуточными между размерами атомов s- и p-элементов данного периода. Изменение радиусов их атомов по периоду более плавное, чем для s- и p-элементов.
В подгруппах d-элементов радиусы атомов в общем увеличиваются. Важно отметить следующую особенность: увеличение атомных и ионных радиусов в подгруппах d-элементов в основном отвечает переходу от элемента 4-го к элементу 5-го периода. Соответствующие же радиусы атомов d-элементов 5-го и 6-го периодов данной подгруппы примерно одинаковы. Это объясняется тем, что увеличение радиусов за счет возрастания числа электронных слоев при переходе от 5-го к 6-му периоду компенсируется f-сжатием, вызванным заполнением электронами 4f-подслоя у f-элементов 6-го периода. В этом случае f-сжатие называется лантаноидным. При аналогичных электронных конфигурациях внешних слоев и примерно одинаковых размерах атомов и ионов для d-элементов 5-го и 6-го периодов данной подгруппы характерна особая близость свойств.
Отмеченным закономерностям не подчиняются элементы подгруппы скандия. Для этой подгруппы типичны закономерности, характерные для соседних подгрупп s-элементов.
50. Получение высокомолекулярных соединений. Реакции полимеризации (ступенчатая и цепная полимеризация).
Природные ысокомолекулярные соединения, образующиеся в клетках живых организмов в результате биосинтеза, могут быть выделены из растительного и животного сырья с помощью экстрагирования, фракционного осаждения и других методов. Основные пути получения синтетических высокомолекулярных соединений - полимеризация иполиконденсация.
Карбоцепные высокомолекулярные
соединения обычно
синтезируют полимеризацией мономеров
по кратным углерод-углеродным связям.
Гетероцепные высокомолекулярные
соединения получают
поликонденсацией, а также полимеризацией
мономеров по кратным гетероатомным
связям типа С=О, N=C—О, С ![]() N
(например, альдегиды, изоцианаты, нитрилы)
или с раскрытием гетероциклических
группировок (например, окисей олефинов,
лактамов).
N
(например, альдегиды, изоцианаты, нитрилы)
или с раскрытием гетероциклических
группировок (например, окисей олефинов,
лактамов).
Полимеризация – реакция образования полимера без образования низкомолекулярных продуктов. В качестве мономера используется молекула, содержащая кратную связь. При полимеризации этилена роль бифункциональной структурной единицы играет двойная связь, которая под влиянием инициатора (например, органического пероксида перикиси бензолоила (C6H5COO)2), легко переходит в радикальное состояние R∙; присоединение радикала создает условия для роста цепи:
|
|
инициирование |
|
|
рост цепи |
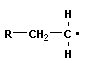
Для реакции полимеризации характерны три стадии: инициирование, рост цепи и обрыв цепи:
|
|
обрыв цепи |
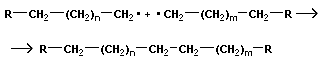
Этот тип полимеризации называется радикальным.
Полимеризация может инициироваться катионами или анионами (ионами). Ионная полимеризация включает те же стадии (инициирование, рост цепи, обрыв цепи). Инициаторами катионной полимеризации могут быть H+, неорганические апротонные кислоты SnCl4, AlCl3, металлоорганические соединения Al(C2H5)3. Инициаторами анионной полимеризации обычно служат электронодонорные соединения (щелочные металлы, их алкоголяты и т. д.).
Катионная полимеризация:
+ |
|
|
|
|
и т. д. |
Анионная полимеризация:
|
|
|
|
|
|
|
|
рост цепи |
|
|
обрыв цепи |
Полимеризация может осуществляться между разными мономерами. Такие соединения называют сополимерами. В табл. 12.9 приведены примеры полимеров и сополимеров, получаемых реакцией полимеризации.
Мономер
|
||||||||||||||||||
Таблица 12.9 Важнейшие полимеры и сополимеры |
Поликонденсация сопровождается образованием полимера и низкомолекулярного соединения (H2O, HCl, NH3 и т. п.). Мономеры должны содержать минимум две функциональные группы.
Типичная реакция поликонденсации лежит в основе получения фенолформальдегидных смол
(n + 2)
|
+ |
n
|
|
|
+ |
n
|
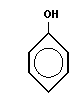
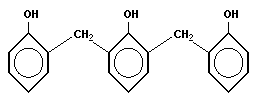
или полиэфирных соединений
![]() +
+
![]()
![]() + n
+ n
В табл. 12.10 приведены наиболее типичные примеры, получаемые реакцией поликонденсации.
Мономеры
|
||||||||||||||||||
Таблица 12.10 Важнейшие полимеры и сополимеры |
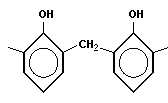
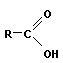 +
+
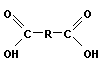
51. Полярные и неполярные молекулы. Дипольный момент связи и дипольный момент молекулы.
ПОЛЯРНЫЕ МОЛЕКУЛЫ, молекулы, обладающие постоянным дипольным моментом в отсутствие внеш. электрич. поля. Дипольный момент присущ таким молекулам, у к-рых распределение электронного и ядерного зарядов не имеет центра симметрии. Обычно полярность отдельных фрагментов молекулы или хим. связей между двумя атомами (или большим числом атомов) определяется величиной соответствующего дипольного момента: чем он больше, тем сильнее полярность.
Под влиянием внеш. электрич. поля в-во поляризуется, т.е. в нем возникает дипольный момент единицы объема. У в-в, состоящих из полярных молекул, поляризация обусловлена смещением электронной плотности под влиянием поля и ориентацией молекул в поле. Ориентации молекул препятствует тепловое движение, поэтому изучение зависимости поляризации от т-ры позволяет определятьдипольный момент молекул (ур-ние Ланжевена-Дебая; см. Диэлектрики). Для двухатомных молекул полярность часто связывают с приближенным представлением электронной волновой ф-ции в рамках валентных связей метода как суммы двух слагаемых, одно из к-рых отвечает ковалентной схеме, другое -ионной валентной схеме. Такое соотнесение позволяет ввести понятие о степени ковалентности или степени ион-ности хим. связи, причем полярность связи определяется в осн. ионной составляющей. Для многоатомных молекул также возможно подобное приближенное выделение в электронной волновой ф-ции ковалентной и ионной составляющих.
Дипольный
момент электрический,
векторная величина, характеризующая
асимметрию распределения положительных
и отрицательных зарядов в электрически
нейтральной системе. Два одинаковых по
величине заряда +qи —q образуют
электрический диполь с дипольный момент
m = q
l, где l -
расстояние между зарядами. Для системы
изn зарядов qi радиусы-векторы
которых ri,  В молекулах и
молекулярных системах центры положительных
зарядов qА совпадают
с положениями атомных
ядер (радиусы-векторы rA),
а электронное распределение описывается
плотностью вероятности r(r).
В молекулах и
молекулярных системах центры положительных
зарядов qА совпадают
с положениями атомных
ядер (радиусы-векторы rA),
а электронное распределение описывается
плотностью вероятности r(r).
В
этом случае дипольный момент 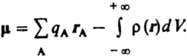 Вектор
дипольный момент направлен от центра
тяжести отрицательных зарядов к центру
тяжести положительных. В хим. литературе
дипольный момент молекулы иногда
приписывают противоположное направление.
Часто вводят представление о дипольный
момент отдельных хим. связей, векторная
сумма которых дает дипольный момент
молекулы. При этом дипольный момент
связи определяют двумя положительными
зарядами ядер атомов, образующих связь,
и распределением отрицательного
(электронного) заряда.
Вектор
дипольный момент направлен от центра
тяжести отрицательных зарядов к центру
тяжести положительных. В хим. литературе
дипольный момент молекулы иногда
приписывают противоположное направление.
Часто вводят представление о дипольный
момент отдельных хим. связей, векторная
сумма которых дает дипольный момент
молекулы. При этом дипольный момент
связи определяют двумя положительными
зарядами ядер атомов, образующих связь,
и распределением отрицательного
(электронного) заряда.
Дипольный момент химической связи обусловлен смещением электронного облака в сторону одного из атомов. Связь называют полярной, если соответствующий дипольный момент существенно отличается от нуля. Возможны случаи, когда отдельные связи в молекуле полярны. а суммарный дипольный момент молекулы равен нулю; такие молекулы наз. неполярными (напр., молекулы СО2 и CCl4). Если же дипольный момент молекулы отличен от нуля, молекула наз. полярной. Напр., молекула Н2О полярна; суммирование дипольных моментов двух полярных связей ОН также дает отличный от нуля дипольный момент, направленный по биссектрисе валентного угла НОН.
Порядок величины дипольный момент молекулы определяется произведением заряда электрона (1,6.10-19 Кл) на длину химической связи (порядка 10-10 м), т. е. составляет 10-29 Кл.м. В справочной литературе дипольный момент молекул приводят в дебаях (Д или D), по имени П. Дебая; 1 Д = 3,33564.10-30 Кл.м.
Спектроскопические методы определения дипольного момента молекул основаны на эффектах расщепления и сдвига спектральных линий в электрическом поле (эффект Штарка). Для линейных молекул и молекул типа симметричного волчка известны точные выражения, связывающие дипольный момент со штарковским расщеплением линийвращательных спектров. Этот метод дает наиб. точные значения величины дипольный момент (до 10-4 Д), причем экспериментально определяется не только величина, но и направление вектора дипольный момент Важно, что точность определения дипольный момент почти не зависит от его абсолютной величины. Это позволило получить весьма точные значения очень малых дипольный момент ряда молекул углеводородов. которые нельзя надежно определить другими методами. Так, дипольный момент пропана равен 0,085 b 0,001 Д, пропилена 0,364 b 0,002 Д, пропина 0,780 b 0,001 Д, толуола 0,375 b 0,01 Д, азулена 0,796 b 0,01 Д. Область применения метода микроволновой спектроскопии ограничена, однако, небольшими молекулами, не содержащими атомов тяжелых элементов. Направление вектора дипольный момент молекулы может быть определено экспериментально и по эффекту Зеемана второго порядка.
Другая группа методов определения дипольных моментов основана на измерениях диэлектрической проницаемости ε вещества. Этими методами измерены дипольные моменты молекул более 10 тыс. веществ. Переход от измеряемого значения ε газа, чистой жидкости или разбавленного раствора, то есть макроскопической характеристики диэлектрика, к величине дипольного момента основан на теории поляризации диэлектриков. Считается, что при наложении электрического поля на диэлектрик его полная поляризация Р (средний дипольный момент единицы объема) складывается из наведенной, или индуцированной, поляризации Рм и ориентационной поляризации Рор и связана с m ур-нием Ланжевена - Дебая:
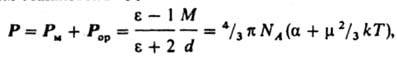
где М - мол. масса, d - плотность, a - поляризуемость молекулы, NA - число Авогадро, k - постоянная Больцмана, Т - абсолютная температура. Измерения диэлектрической проницаемости проводят в постоянном поле или при низких частотах, обеспечивающих полную ориентацию молекул по полю. При наиболее распространенном варианте метода - измерениях в разбавленных растворах неполярных растворителей - предполагается аддитивность поляризаций растворенного вещества и растворителя.
Сопоставление дипольных моментов полярных молекул некоторых органических соединений, полученных разными методами, показано в таблице.
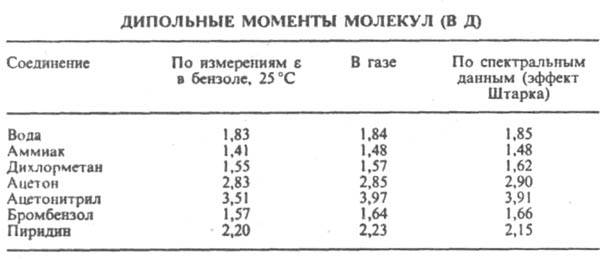
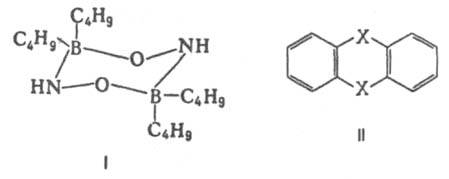
Важнейшая область применения данных о дипольных моментах молекул - структурные исследования, установление конформации молекул, конформационного и изомерного состава вещества, его зависимости от температуры. Величины дипольного момента молекул позволяют судить о распределении электронной плотности в молекулах и зависимости этого распределения от характера отдельных заместителей. В общем случае структурная интерпретация дипольный момент требует сравнения экспериментальных величин со значениями, полученными квантово-механическим расчетом либо при помощи аддитивной векторной схемы с использованием дипольных моментов отдельных связей и атомных групп. Последние находят либо по интенсивностям колебательных полос поглощения, либо путем векторного разложения дипольный момент некоторых симметричных молекул. Расчеты с использованием векторной аддитивной схемы могут учитывать различные проявления стереохимической нежесткости, например, затрудненное или свободной внутреннее вращение молекулы. Высокосимметричные молекулярные структуры, обладающие центром инверсии, двумя взаимно перпендикулярными осями вращения или осями, перпендикулярными плоскости симметрии, не должны иметь дипольный момент. По наличию или отсутствию дипольного момента молекулы можно в отдельных случаях выбрать для нее ту или иную структуру без каких-либо теоретических расчетов. Так, равенство нулю экспериментального дипольный момент димера аминооксидибутилборана (формула I) служит доказательством того, что он существует в виде устойчивой кресловидной конформации, обладающей центром инверсии. Наоборот, наличие дипольный момент у тиантрена (формула II, X = S) и селенантрена (II, X = Se), равных 1,57 Д и 1,41 Д соотв., исключает для них центросимметричную структуру, в частности плоскую.
52 – смотри 72
53. Понятие об электронных потенциалах. Уравнение Нернста.
Электродный потенциал, разность электрических потенциалов между электродом и находящимся с ним в контакте электролитом(чаще всего между металлом и раствором электролита). Возникновение Электродный потенциал обусловливается переносом заряженных частиц через границу раздела фаз, специфической адсорбцией ионов, а при наличии полярных молекул (в том числе молекул растворителя) — ориентационной адсорбцией их. Величина Электродный потенциал в неравновесном состоянии зависит как от природы и состава контактирующих фаз, так и от кинетических закономерностей электродных реакций на границе раздела фаз. Равновесное значение скачка потенциалов на границе раздела электрод/раствор определяется исключительно особенностями электродной реакции и не зависит от природы электрода и адсорбции на нём поверхностно-активных веществ. Эту абсолютную разность потенциалов между точками, находящимися в двух разных фазах, нельзя измерить экспериментально или рассчитать теоретически. Практическое значение имеют относительные Электродный потенциал, обычно называемые просто Электродный потенциал, представляющие собой разность Электродный потенциал рассматриваемого электрода и электрода сравнения — чаще всего нормального водородного электрода, Электродный потенциал которого условно принимается равным нулю. При электрохимическом равновесии на электроде величина Электродный потенциал (E) может быть выражена через изменениегиббсовой энергии (DG) реакции: Е = —DG/zF, где z — число электронов, участвующих в электрохимическом процессе, F — Фарадея число. Электродный потенциал в этом случае зависит от активности (а) участвующих в реакции веществ (потенциалопределяющих веществ). Для электродов Me/Men+ Е = E0 + (RT/zF) ln aMen+, где R — газовая постоянная, Т —температура, E0 — нормальный потенциал. Для окислительно-восстановительных систем с инертным электродом, у которых все компоненты электрохимической реакции находятся в растворе, Электродный потенциал (окислительно-восстановительный потенциал) определяется активностями как окисленной (aok), так и восстановленной (ав) форм вещества.
Уравнение
Нернста:
![]() ,
где n — стехиометрический коэффициент.
В случае, когда на электроде возможно
одновременное протекание более одной
электродной реакции, используется
понятие стационарного Электродный
потенциал При
пропускании электрического тока
измеренный Электродный
потенциал будет
отличаться от равновесного на величину
поляризации (см. Поляризация
электрохимическая).
,
где n — стехиометрический коэффициент.
В случае, когда на электроде возможно
одновременное протекание более одной
электродной реакции, используется
понятие стационарного Электродный
потенциал При
пропускании электрического тока
измеренный Электродный
потенциал будет
отличаться от равновесного на величину
поляризации (см. Поляризация
электрохимическая).
![]() ,
,
где ![]() — универсальная
газовая постоянная,
равная 8.31 Дж/(моль·K);
— универсальная
газовая постоянная,
равная 8.31 Дж/(моль·K);
![]() —
абсолютная
температура;
—
абсолютная
температура;
![]() — число
Фарадея,
равное 96485,35 Кл/моль;
— число
Фарадея,
равное 96485,35 Кл/моль;
![]() —
число молей электронов,
участвующих в процессе;
—
число молей электронов,
участвующих в процессе;
![]() и
и ![]() — активности соответственно окисленной и восстановленной форм
вещества, участвующего в полуреакции.
— активности соответственно окисленной и восстановленной форм
вещества, участвующего в полуреакции.
54. Последовательность заполнения электронных оболочек атомов. Принцип наименьших энергий. Принцип Паули. Правило Хунда.
Правило Хунда(Гунда) определяет порядок заполнения орбиталей определённого подслоя и формулируется следующим образом: суммарное значение спинового квантового числа электронов данного подслоя должно быть максимальным.
Это означает, что в каждой из орбиталей подслоя заполняется сначала один электрон, а только после исчерпания незаполненных орбиталей на эту орбиталь добавляется второй электрон. При этом на одной орбитали находятся два электрона с полуцелыми спинами противоположного знака, которые спариваются (образуют двухэлектронное облако) и, в результате, суммарный спин орбитали становится равным нулю.
При́нцип Па́ули (принцип запрета) — один из фундаментальных принципов квантовой механики, согласно которому два и более тождественных фермиона не могут одновременно находиться в одном квантовом состоянии.
Принцип был сформулирован для электронов Вольфгангом Паули в 1925 г. в процессе работы над квантомеханической интерпретациейаномального эффекта Зеемана и в дальнейшем распространён на все частицы с полуцелым спином. Полное обобщённое доказательство принципа было сделано им в 1940 г. в рамках релятивистской квантовой механики: волновая функция системы фермионов являетсяантисимметричной относительно их перестановок, поведение систем таких частиц описывается статистикой Ферми — Дирака.
Принцип Паули можно сформулировать следующим образом: в пределах одной квантовой системы в данном квантовом состоянии может находиться только одна частица, состояние другой должно отличаться хотя бы одним квантовым числом.
В статистической физике принцип Паули иногда формулируется в терминах чисел заполнения: в системе одинаковых частиц, описываемых антисимметричной волновой функцией, числа заполнения могут принимать лишь два значения Np = 0,1
55. Превращение химической энергии в электрическую.
Гальвани́ческий элеме́нт — химический источник электрического тока, названный в честь Луиджи Гальвани. Принцип действия гальванического элемента основан на взаимодействии двух металлов через электролит, приводящем к возникновению в замкнутой цепи электрического тока. ЭДС гальванического элемента зависит от материала электродов и состава электролита. Сейчас широко распространены следующие гальванические элементы:
Тип |
ЭДС (В) |
Достоинства |
угольно-цинковые (солевые) |
1,5 |
дешёвые |
щелочные (жаргонное название — алкалиновые) |
1,6 |
высокий ток, ёмкие |
никельоксигидроксидные (NiOOH) |
1,6 |
высокий ток,очень ёмкие |
литиевые |
3,0 |
очень высокий ток, очень ёмкие |
Распространены солевые и щелочные элементы следующих типоразмеров:
Американское название |
Название МЭК |
Название ГОСТ |
Обиходное название |
AAAA |
R61 |
???? |
???? |
AAA |
R03 |
286 |
мизинчик, тонкая |
AA |
R6 |
316 |
пальчик |
C |
R14 |
343 |
дюймовочка |
D |
R20 |
373 |
большая, бочка |
Распространены солевые и щелочные батареи элементов следующих типоразмеров:
Название МЭК |
Название ГОСТ |
Обиходное название |
Описание |
3R12 |
3336 |
квадратная, плоская |
3 элемента 12 (337) 4,5 В |
6LR61 |
_ |
крона |
6 спец. галетных элементов 9 В |
В названии МЭК для щелочных элементов перед буквой R добавляется L, а для никельоксигидроксидных - буква X.
Также известны несколько десятков типоразмеров пуговичных (таблеточных) элементов разных электрохимических систем. Их обычно применяют в часах, микрокалькуляторах и других малогабаритных устройствах.
56. Предельные углеводороды. (см.46). Изомеры и гомологи.
Изомеры (от др.-греч. ἴσος — «равный», и μέρος — «доля, часть») — соединения (главным образом органические), одинаковые по элементному составу и молекулярной массе, но различные по физическим и химическим свойствам.
Гомологический ряд — ряд химических соединений одного структурного типа (например, алканы или алифатическиеспирты — спирты жирного ряда), отличающихся друг от друга по составу на определенное число повторяющихся структурных единиц — т. н. «гомологическую разность». Чаще всего это метиленовые звенья: …—СН2—… Простейший пример гомологического ряда — низшие гомологи алканов (общая формула СnH2n+2): метан CH4, этан C2H6, пропан С3H8и т. д.
57. Растворы полимеров. Растворимость и набухание полимеров.
Растворы полимеров
РАСТВОРЫ ПОЛИМЕРОВ
,
обладают рядом особенностей по сравнению с р-рами низкомол. в-в из-за св-в макромолекул: больших размеров, широкого диапазона гибкости (жесткости), большого набора конформаций, способности к конформац. перестройкам при изменении т-ры, р-рителя и т. п.
![]()
В разбавленных Р.
п. гибкоцепные синтетич. макромолекулы
принимают конформацию статистич. клубка,
в объеме к-рого концентрация с 0 собств.
звеньев весьма мала (порядка 1% по массе
и менее) и убывает с ростом мол. массы.
Влияние мол. массы и геом. характеристик
макромолекул на св-ва Р. п. приводит к
тому, что концентрация растворенного
в-ва не является однозначным критерием
разделения Р. п. на разбавленные и
концентрированные, в отличие от р-ров
низкомол. в-в. Р. п. со средней концентрацией
срастворенного в-ва считается разбавленным,
если с
< с 0,
т. е. среднее расстояние между
макромолекулами значительно больше их
размера. Условный показатель,
характеризующий уд. объем, занимаемый
макромолекулой,-характеристическая
вязкость, где h и h0 -соотв.
вязкость р-ра с концентрацией с и
вязкость чистого р-рителя. Величина
[h]-условный показатель, определяющий
прирост вязкости р-рителя при введении
в него полимера. Если концентрация Р.
п. выражена в г/л, то единица характеристич.
вязкости-л/г или м 3/кг.
Р. п. является "разбавленным", если
[h]c <
1. Разбавленные Р. п. используют для
изучения характеристик индивидуальных
макромолекул: мол. массы, размеров,
конформаций, гибкости цепи и т. п. Интервал
концентраций ![]() определяет
т. наз. полуразбавленный Р. п.: концентрация
полимера мала, но статистич. клубки
перекрываются, проникая друг в друга.
Клубковые кон-формации макромолекул
сохраняются и в концентрированных Р.
п., а также в свсрхконцентрированных Р.
п., примером к-рых служат полимеры,
пластифицированные труднолетучими
р-рителями (см. Пластификация
полимеров).
определяет
т. наз. полуразбавленный Р. п.: концентрация
полимера мала, но статистич. клубки
перекрываются, проникая друг в друга.
Клубковые кон-формации макромолекул
сохраняются и в концентрированных Р.
п., а также в свсрхконцентрированных Р.
п., примером к-рых служат полимеры,
пластифицированные труднолетучими
р-рителями (см. Пластификация
полимеров).
Др. особенность Р. п.-понижение совместимости компонентов из-за того, что энтропия смешения DS с снижается при соединении мономеров в единую цепь. Полное смешение полимеров с низкомол. р-рителями возможно лишь в определенном интервале т-р. Вне этого интервала взаимная р-римость компонентов становится ограниченной и происходит разделение Р. п. на две фазы, сосуществующие в равновесии.
На гипотетич. обобщенной диаграмме р-римости в координатах концентрация полимера с-т-ра T (рис. 1) имеется кривая (бинодаль), отделяющая замкнутую область т-р и концентраций, внутри к-рой Р. п. расслаивается на две фазы. Максимум и минимум на бинодали определяют верхнюю и нижнюю критич. т-ры смешения (соотв. ВКТС и НКТС) и критич. концентрации. В критич. точках составы сосуществующих фаз полимер - р-ритель совпадают. При т-рах вне интервала ВКТС-НКТС имеет место неограниченное взаимное смешение компонентов, причем ниже НКТС - за счет сильного взаимод. активных атомных групп полимера и р-рителя, напр. за счет водородных связей. Вблизи критич. т-ры перехода жидкость-пар (НКТС') также имеется область ограниченной совместимости.
Реальная полная диаграмма р-римости полимера с тремя критич. т-рами до сих пор не получена. Наиб. часто проявляется на практике лишь ВКТС (хотя она может лежать выше точки кипения р-рителя), а НКТС не достигается, чаще всего из-за высокой т-ры замерзания р-рителя. Имеются и системы, в к-рых существует лишь НКТС.
Форма и положение бинодали для данной пары полимер -р-ритель зависит от мол. массы. С ростом мол. массы М полимера бинодаль смещается в сторону малых концентраций и больших т-р, критич. концентрация с кр~ 1/М 1/2, 1/Т кр линейно убывает с убыванием 1/М 1/2 (пунктирная линия на рис. 1), т. е. р-римость полимера уменьшается. Такая зависимость используется для фракционирования полимеров по мол. массе. Предельное значение Т кр при М : со определяет т. наз. 9-температуру Флори (q-точку, q-усло-вие) как идеальную т-ру, при к-рой клубкообразные макромолекулы в р-ре имеют конформацию гауссова клубка, т. е. их средний квадратичный линейный размер Rпропорционален М 1/2, а с 0 ~ 1/М 1/2. С ростом т-ры размеры макромолекулы в разб. р-рах увеличиваются до R~ М 0,6, с 0 ~ М -0,8 (р-ритель "улучшается"). Предельная концентрация с 0 убывает с ростом мол. массы полимера, а при данной мол. массе-с ростом т-ры.
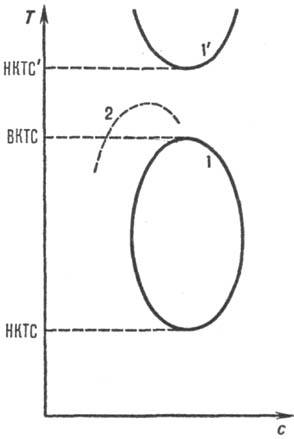
Рис. 1. Обобщенная диаграмма р-римости для системы аморфный полимерЧ р-ритель. На осях отложены концентрация полимера си т-ра Т. Кривые 1 и Г Ч бинодали, ВКТС, НКТС и НКТС'-соотв. верхняя и две нижние критич. т-ры смешения; пунктирная кривая 2 показывает смещение бинодали 1 при увеличении мол. массы полимера.
Теория, позволяющая определить пределы совместимости полимера и р-рителя в зависимости от мол. массы растворенного в-ва и т-ры, развита П. Дж. Флори и М. Хаггинсом в 40-х гг. 20 в. Энтропия смешения D<sc</s системы полимер-р-ритель рассчитывалась на основе решеточной модели (см. Жидкость),согласно к-рой жидкость м. б. представлена квазирешеткой, в каждой ячейке к-рой может помещаться либо молекула р-рителя, либо равный ей по размерам участок макромолекулы, что связано с конкретной конфор-мацией цепи. Соответственно при расчете энтропии смешения D<sc</s принимаются во внимание лишь допустимые кон-формации, а при малой молярной доле полимера в р-ре возможны большие значения D<sc</s. Наличие отличной от нуля теплоты смешения DH с обусловлено тем, что энергия межмол. взаимод. в чистых в-вах-р-рителе и полимереЧ отличается от энергии взаимод. р-ритель-полимер. Избыточная своб. энергия взаимод., приходящаяся на одну молекулу р-рителя в Р. п., характеризуется полуэмпирич. параметром Флори-Хаггинса c,к-рый для данной пары полимер - р-ритель является ф-цией т-ры. С параметром X связан второй вириальный коэф. А 2 разложения осмотич. давления р-ра в ряд по концентрации, позволяющий оценить "качество" р-рителя. Если контакты полимер - полимер и р-ритель - р-ритель энергетически менее выгодны, чем контакты полимер-р-ритель, коэф. А 2 >0, р-ритель считается хорошим. Р-рители с А 2 <0 считаются плохими, р-ри-мость полимера в них ограничена. При А 2= 0 р-ритель наз. квазиидеальным или q-растворителем, а т-ра, при к-рой положит. энтальпия смешения компенсируется возрастанием энтропии, является q-точкой. Для Р. п. q-точка аналогична точке Бойля для реальных газов (см.Вириальное уравнение). Для жесткоцепных полимеров (напр., при длине жесткого сегмента 10 нм и выше) наблюдается ухудшение р-римости сравнительно с гибкоцепными полимерами, совместимость компонентов Р. п. часто достигается за счет сильных взаимод. полимер-р-ритель. Р-римость полимера повышается при наличии у макромолекул подвижных боковых групп атомов. Р-ры жесткоцепных полимеров изотропны лишь при концентрации ниже нек-рой критической, при повышении концентрации на диаграмме р-римости наблюдается узкая область двухфазного состояния, а затем состояние полимерного жидкого кристалла(рис. 2). В изотропных Р. п. вязкость сильно возрастает с концентрацией, в жидких кристаллах макромолекулы ориентационно упорядочены, что обеспечивает уменьшение вязкости. Рис. 2. Равновесная кривая сосуществования фаз в системе полимер ^р-ритель. По осям отложены концентрация полимера си т-ра Т. Буквами И и А обозначены области существования изотропного и анизотропного р-ров соответственно.
Биол. макромолекулы (белки, нуклеиновые к-ты) и их модели (полипептиды, полинуклеотиды) в р-рах могут иметь специфич. конформации, стабилизированные внутри-мол. взаимодействием . Так, нативные глобулярные конформации белков в водном р-рителе стабилизированы водородными связями и гидрофобными взаимодействиями неполярных групп атомов. Полярные группы на пов-сти глобулы обеспечивают ее р-римость. При изменении состава и св-в р-рителя, рН и ионной силы р-ра или при изменении т-ры происходят внутримол. конформац. переходы типа спираль-клубок и глобула-клубок, что приводит к резкому изменению всех св-в Р. п.
Набухание полимеров. Процесс растворения полимеров, как указывалось, проходит через стадию их набухания. Внешне процесс набухания выражается в изменении объема и веса образца вследствие поглощения полимером растворителя. Набухание можно рассматривать как одностороннее смешение, т. е. только как проникание растворителя в полимер. Подвижность макромолекул слишком мала, а силы когезии велики, поэтому вначале макромолекулы полимера не диффундируют в растворитель. Молекулы растворителя, диффундируя в полимер, вначале заполняют в нем межмолекулярные пространства, а затем, по мере увеличения объема растворителя в полимере, начинают раздвигать макромолекулы. Скорость диффузии растворителя в полимер зависит от свойств растворителя и структуры полимера. С увеличением количества продиффундировавшего в полимер растворителя расстояние между макромолекулами постепенно возрастает, что приводит к пропорциональному увеличению размеров набухающего образца. Таким образом, набуханием называют проникание молекул растворителя между макромолекулами полимера, вследствие чего увеличиваются расстояния между отдельными сегментами, а затем и цепями полимера.[
58. Реакции поликонденсации. Фенолформальдегидная смола.
Поликонденсация — процесс синтеза полимеров из полифункциональных (чаще всего бифункциональных) соединений, обычно сопровождающийся выделением низкомолекулярных побочных продуктов (воды, спиртов и т. п.) при взаимодействии функциональных групп.
Молекулярная масса полимера, образовавшегося в процессе поликонденсации, зависит от соотношения исходных компонентов, условий проведения реакции.
В реакции поликонденсации могут вступать как один мономер с двумя различными функциональными группами: например, синтез поли-ε-капроамида (найлона-6, капрон) из ε-аминокапроновой кислоты, так и два мономера, несущие различные функциональные группы, например, синтез найлона-66 поликонденсацией адипиновой кислоты игексаметилендиамина; при этом образуются полимеры линейного строения (линейная поликонденсация, см. Рис.1). В случае, если мономер (или мономеры) несут более двух функциональных групп, образуются сшитые полимеры трёхмерной сетчатой структуры (трёхмерная поликонденсация). С целью получения таких полимеров к смеси мономеров нередко добавляют «сшивающие» полифункциональные компоненты.
Особняком стоят реакции синтеза полимеров из циклических мономеров по механизму раскрытия цикла — присоединение, например, синтез найлона-6 из капролактама(циклического амида ε-аминокапроновой кислоты); несмотря на то, что выделение низкомолекулярного фрагмента при этом не происходит, такие реакции чаще относят к поликонденсации.
Фенолформальдегидная смола (PF) — синтетическая смола, продукт поликонденсации фенола с формальдегидом. Реакция проводится в присутствии кислых (соляная, серная, щавелевая и другие кислоты) или щелочныхкатализаторов (аммиак, гидроксид натрия, гидроксид бария, триэтиламин).
Применяются для получения пластических масс (отвержденные смолы называют резитами, отвержденные в присутствии нефтяных сульфокислот — карболитами, молочной кислоты — неолейкоритами), синтетических клеев,лаков, выключателей, тормозных накладок, подшипников, так же широко используется в изготовление шаров для бильярда.
59 см 50, Промышленные методы получения полимеров.