

МиНистерсво здравоохранения и социального развития рф
Кафедра педиатрии
Учебно-методическое пособие
для студентов педиатрических факультетов, интернов, ординаторов и врачей педиатров.
ЭНДОКРИННЫЕ БОЛЕЗНИ У НОВОРОЖДЁННЫХ
НАРУШЕНИЯ ПОЛОВОЙ ДИФФЕРЕНЦИРОВКИ У НОВОРОЖДЁННЫХ
Пол каждого человека в процессе развития и созревания определяется 4-мя составляющими: генетическим полом (набором половых хромосом), гонадным полом (строением гонад и внутренних половых органов), фенотипическим полом (генитальным, «паспортным» - строением наружных половых органов), психо-сексуальной ориентацией данного индивидуума. Эти 4 пола обычно по мере развития и созревания последовательно дополняют друг друга, а любые дефекты в механизме формирования каждого из них приводят к множеству вариантов нарушения формирования пола в целом. Дефекты первых трёх компонентов нередко проявляются уже в периоде новорождённости.
Регуляция и механизмы половой дифференцировки.
Зачаток гонад определяется у плода на 5-6 неделе беременности в виде половых бугорков. Вначале эти примитивные гонады бипотенциальны и состоят из коркового слоя (cortex), ткани будущих тестикул и мозгового (medulla) – ткань яичников (рис. 1). В них определяются следующие типы клеток: а) герминативные, предназначенные для формиро-вания сперматогоний у мужской особи и ооцитов – у женской; б) опорные клетки, из кото-рых в дальнейшем образуются клетки Сертоли у мужчин и фолликулярные клетки – у женщин; в) стероидные клетки-предшественники клеток Лейдига у мужчин и тека-клеток – у женщин. И хотя концепция будущего пола ребёнка определяется набором половых хромосом, критическим моментом её реализации является дифференцировка опорных клеток в клетки Сертоли.
Генетический мужской пол определяется наличием Y хромосомы, точнее гена, локализованного в коротком плече этой хромосомы, так называемого тестикуло-определяющего фактора. Этот ген выявляется у всех 46XY мужчин, 46ХХ мужчин и не найден у 46XY женщин. Это так называемый ген SRY (Sex-determing Region of Y chromosome – пол-детерминирующий регион Y хромосомы). SRY является транскрипционным фактором для связанного с ним гена SOX-9, который и детерминирует дифференцировку опорных клеток в клетки Сертоли. В свою очередь, клетки Сертоли яичек стимулируют продукцию клетками Лейдига тестостерона и мюллеровского ингибирующего фактора (МИФ), которые способствуют развитию вольфовых структур и регрессию – мюллеровых и формируют мужской фенотип. Третий ген, DAX-1, является антагонистом SRY и подавляет экспрессию гена SOX-9. Клетки Лейдига яичек стимулируются также хорионическим гонадотропином (ХГТ) и вырабатывают значительные количества тестостерона у плода мужского пола, который способствует дифференцировке вольфовых структур в семенные канальцы и эпидидимис. В тканях тестостерон локально превращается под действием фермента 5a-редуктазы в дигидротестостерон (ДГТ). Именно ДГТ определяет развитие полового члена и мошонки. Развитие мужских гениталий завершается к 12-й неделе беременности. Во 2-м триместре беременности под действием МИФ происходит абдоминальное опущение яичек, а их дальнейшее продвижение в мошонку и рост полового члена определяются во 2-ом и 3-ем триместрах повышенной продукцией тестостерона под действием гонадотропных гормонов, продуцируемых гипофизом плода.
При генотипе 46ХХ и отсутствии генов SRY или SOX-9 к 10-й неделе беременности формируются яичники с последующим формированием наружных половых органов женского типа без каких-либо гормональных влияний (фетальные яичники у плода женского пола интактны) вследствие так называемой автономной тенденции плода к феминизации.
Дефекты формирования генетического пола.
Набор половых хромосом определяет дифференцировку гонад и формирование их по мужскому или женскому типу. К настоящему времени описано множество различных вариантов хромосомных нарушений, некоторые из них приводят к гибели плода (Y0), другие сопровождаются патологией развития гонад и наружных половых органов (X0), гормонально-метаболическими и соматическими аномалиями (XXX). В отличие от аутосом лишние Х хромосомы не вызывают значительных патологических изменений, т.к. все хромосомы кроме одной неактивны и не требуются для использования в общем пуле генетической информации. С другой стороны, для нормального формирования гонад необходимо не менее 2-х Х хромосом.
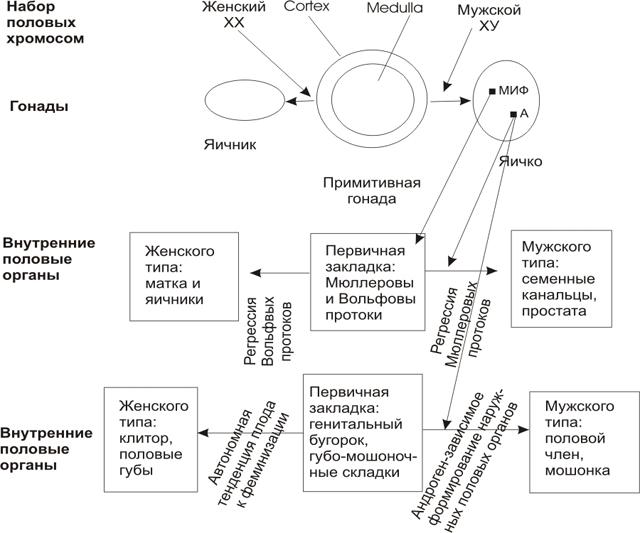
рис. 1. Схема регуляции процессов половой дифференцировки.
А – андрогены; МИФ – мюллеров ингибирующий фактор; (см. пояснения в тексте).
Встречаются и сложные для их понимания варианты хромосомных аномалий. Для формирования тестикул и мужского фенотипа необходима Y хромосома, однако описаны случаи истинного гермафродитизма с мужским фенотипом и с женским набором хромосом ХХ. Этиология таких изменений остаётся неясной, т.к. в большинстве случаев истинного гермафродитизма ген SRY не обнаруживается.
Наиболее часто встречающиеся «классические» варианты нарушения расхождения половых хромосом определяются уже при рождении и в периоде новорождённости. Частота таких аномалий у новорождённых составляет: XXY – 1:545, XYY – 1:728, XXX – 1:727, 45 X0 – 1:2181. При меньшей частоте распространённости синдром Шерешевского-Тернера (45 X0) диагностируется чаще других, т.к. проявляется и разнообразными соматическими аномалиями (стигмами дисэмбриогенеза), а остальные из вышеприведенных синдромов в периоде новорождённости обычно не выявляются.
Синдром Шерешевского-Тернера.
Классическим вариантом этого синдрома является кариотип 45 Х0 – 50% всех случаев. Более редко встречаются аномалии одной из Х хромосом: делеция части Х хромосомы – 46 X,i (Xq, изохромосома) – 17%, кольцевая Х хромосома (Xr) и другие дефекты. Встречается и так называемый «мозаицизм», когда часть клеточного клона содержит набор 46 ХХ, а другая Х0 (45Х0/46XY) – 8%, реже выявляется сочетание 45 Х0 с 46 XY и другие варианты. Истинная частота формирования набора 45 Х0, по-видимому, более высокая, но срабатывает механизм защиты генофонда, т.к. в случае такой аномалии нередко происходит выкидыш до 28-й недели беременности, при этом частота выявления кариотипа 45 Х0 среди спонтанных абортов составляет 1:15.
При мозаичном варианте синдрома наличие в части клеток нормального набора половых хромосом 46 ХХ уменьшает степень выраженности соматических аномалий (отсутствуют крыловидные складки), в 12% таких случаев возможно развитие матки и яичников и в дальнейшем нормальное половое созревание.
С другой стороны, если при мозаицизме имеется Y хромосома (X0/XY или XX/XY), или выявлен ген SRY, то в закладке гонад могут развиться и элементы тестикул; наружные половые органы могут быть мужского или женского типа, гермафродитными, женскими с гипертрофией клитора, мужскими с гипоспадией и крипторхизмом. При наличии Y хромосомы наиболее часто встречается мужской фенотип с опустившимися тестикулами, что было подтверждено пренатальным исследованием кариотипа в амниотической жидкости. Такой тип мозаицизма носит название «смешанный гонадальный дисгенез», при этом нередко в закладке гонад может происходить злокачественная дегенерация их ткани, особенно при внутрибрюшном расположении гонад и женском фенотипе ребёнка, поэтому в таких случаях показана ранняя гонадэктомия.
К типичным проявлениям синдрома Шерешевского-Тернера относятся: короткая шея с крыловидными складками (у новорождённых они выглядят как дополнительные складки на задней поверхности шеи); «треугольное» лицо с низко расположенными ушными раковинами; готическое нёбо; низкое расположение волос на затылке, широкая и развёрнутая, «щитообразная», грудная клетка с широко расположенными и гипопластичными сосками; разболтанность суставов. Среди других, непостоянно встречающихся, аномалий можно отметить коарктацию аорты, бикуспидальный аортальный клапан, гемангиомы, cutis laxa, пигментные невусы, диспластичные ногти, склонность к формированию келоидных рубцов, лимфедема на дорзальной поверхности кистей и стоп, клювовидный медиальный мыщелок большеберцовой кости, дистальные фаланги кистей рук в виде «барабанных палочек», укорочение метакарпальных или метатарзальных костей, изменения дерматоглифики. Характерны также частые повторные средние отиты, присоединение аутоиммунного лимфоцитарного тиреоидита, идиопатическая гипертензия. Основными проблемами в более старшем возрасте являются низкорослость и первичный (гипергонадотропный) гипогонадизм.
Дефекты формирования гонадного пола.
Аномалии расхождения половых хромосом или дефекты их структуры обычно сопровождаются нарушениями развития гонад, их дисгенезией. В литературе описаны случаи подобных дисгенезий, при которых хромосомные аберрации не были найдены и этиология которых неизвестна. В ряде случаев удаётся выявить воздействие тератогенных факторов, оказывавших повреждающее действие на формирующиеся гонады, такие как радиация, вирусы, лекарственные препараты и различные токсические вещества. В зависимости от времени и степени воздействия этих факторов на гонады (прежде всего на тестикулы) тяжесть повреждения строения внутренних и наружных половых органов может быть различной.
Чистый гонадальный дисгенез. Нарушений расхождения половых хромосом нет, кариотип или чисто мужской, или чисто женский. Вследствие полной дисгенезии гонад и «тенденции плода к феминизации» (при исключении всех гормональных влияний наружные половые органы плода формируются как женские), характерен нормальный женский фенотип.
Девочки высокорослые, с евнухоидными пропорциями тела, вторичные половые признаки отсутствуют, наружные половые органы инфантильны, характерна первичная аменорея. При кариотипе 46XX возможен аутосомно-рецессивный тип наследования и нейро-сенсорный тип глухоты. Вариант кариотипа 46 XY также наследуется по аутосомно-рецессивному типу, но может передаваться и как связанная с Х хромосомой генная мутация; нередко выявляется случайно в неонатальном периоде.
Частичный гонадальный дисгенез. Этот вариант дисгенезии обычно развивается, если тератогенные факторы оказывали своё действие на поздних стадиях развития плода и характеризуется значительным клиническим полиморфизмом.
Повреждение тестикул между 9-й и 12-й неделями беременности не нарушает инволюцию Мюллеровых протоков, т.к. МИФ уже секретировался и оказал своё действие на характер развития внутренних половых органов, но формирование наружных половых органов нарушается. Нормальное формирование последних по мужскому типу требует достаточной продукции тестостерона для преодоления тенденции к феминизации. Наружные половые органы при этом женского типа, а гонады, матка и фаллопиевы трубы отсутствуют.
Если тестикулы повреждаются во 2-м триместре беременности (синдром исчезнувших тестикул) наружные и внутренние половые органы формируются по мужскому типу, но выявляется врождённая анорхия. При менее значительном повреждении гонад возможно развитие синдрома микропениса или крипторхизма.
Истинный гермафродитизм. В классическом варианте у одного и того же индивидуума часть клеток содержит набор половых хромосом 46ХХ, а другая – 46XY. При этом одновременно формируется как ткань яичка, так и ткань яичников, что создаёт различные варианты строения гонад: с одной стороны яичко, а с другой – яичник; с одной стороны яичко или яичник, а с другой овотестис (единая гонада, часть ткани которой составляют элементы тестикул, а другая – яичника); овотестис с обеих сторон.
В 60-70% всех случаев при истинном гермафродитизме выявляется кариотип 46XХ, в остальных случаях 46ХY. Кариотип 46 ХХ приблизительно в 50% случаев ассоциируется с H-Y антигеном и лишь в небольшом проценте случаев выявляется ген SRY. Предполагают, что при истинном гермафродитизме с кариотипом 46ХХ возможна аутосомная или связанная с Х хромосомой мутация гена, контролирующего работу SRY, играющего важную роль в дифференцировке гонад.
Особенности дифференцировки как внутренних, так и наружных половых органов зависят от степени морфо-функционального развития тестикулярной ткани. Тип дифференцировки протоков определяется типом гонады на каждой из сторон, так как МИФ действует локально. Наружные половые органы при истинном гермафродитизме всегда (по крайней мере, у человека) имеют неопределённое, гермафродитное строение.
Дефекты формирования генитального пола.
В классических случаях речь идёт о вариантах неправильного строения наружных половых органов, которые не связаны с генетическими дефектами или аномалиями развития гонад. Такие состояния называют ложным гермафродитизмом или псевдогермафродитизмом. При этом наружные половые органы могут быть мужского типа с недостаточной андрогенизацией или женского типа с андрогенизацией. Нарушения формирования наружных половых органов при нормальном кариотипе и нормально развитых гонадах являются следствием воздействия тератогенных факторов в эмбриогенезе, первично генетических дефектов промежуточных механизмов эмбриогенеза, генетических дефектов различных рецепторов или пострецепторных механизмов передачи действия гормонов.
Наружные половые органы могут быть полностью или частично гермафродитными, могут быть нормально сформированы, но не соответствовать генетическому и/или гонадному полу ребёнка. Поскольку по строению наружных половых органов невозможно классифицировать подобные состояния, то в зависимости от строения внутренних половых органов выделяют мужской и женский варианты ложного гермафродитизма.
Ложный женский гермафродитизм. Андрогенизация наружных половых органов плода женского пола (по набору половых хромосом и строению гонад) происходит в результате повышенной продукции андрогенов или в организме самого плода, или при их избыточном поступлении из организма матери. Если это происходит до 12-й недели внутриутробного развития, в самом начале формирования наружных половых органов, то происходит формирование урогенитального синуса (уретра и влагалище открываются одним отверстием), большие половые губы или не изменяются, или напоминают расщеплённую мошонку, иногда одновременно происходит атрезия наружных 2/3 влагалища. После 12-й недели избыток андрогенов приводит только к увеличению клитора разной степени выраженности.
В р о ж д ё н н а я г и п е р п л а з и я к о р ы н а д п о ч е ч н и к о в. Один из наиболее частых вариантов гермафродитизма – 25% всех случаев. Это наследственно-обусловленная болезнь, в основе развития которой лежит недостаточность ферментов (более чем в 90% случаев 21- и 11-гидроксилаз), участвующих в биосинтезе глюкокортикоидов и минералокортикоидов в коре надпочечников. При этом синтез андрогенов в надпочечниках не нарушается, а повышенная продукция АКТГ (по принципу обратной связи в ответ на недостаточный синтез глюкокортикоидов) приводит к повышенной продукции надпочечниками плода андрогенов и «врождённой гиперплазии надпочечников» (общепринятое название болезни в зарубежной литературе). Гермафродитное строение гениталий с андрогенизацией различной степени выраженности отмечается только у девочек, генетический пол и внутренние половые органы нормально сформированы, женского типа, независимо от степени андрогенизации наружных половых органов. У мальчиков отмечаются различной степени выраженности макрогенитосомия (увеличение размеров наружных половых органов) и их пигментация.
При незначительной андрогенизации у девочек (только небольшое увеличение клитора) и нередко у мальчиков при рождении изменения в строении наружных половых органов не замечают. В этом случае при полной недостаточности фермента 21-гидроксилазы (тяжёлая, сольтеряющая форма болезни) на 2-3-и сутки после рождения, когда организм новорождённого начинает обеспечиваться только собственными гормонами надпочечников, появляются признаки острой надпочечниковой недостаточности с обезвоживанием, синдромом потери соли. Две другие формы болезни – простая, вирильная или компенсированная (частичная недостаточность 21-гидроксилазы) и гипертоническая (недостаточность 11-гидроксилазы) не сопровождаются симптомами надпочечниковой недостаточности. Если они не выявляются при рождении, то будут диагностированы самое раннее к концу 1-го года жизни, а чаще в возрасте 2-4 лет, в связи с появлением признаков преждевременного полового созревания у мальчиков по гомосексуальному, а у девочек по гетеросексуальному типу.
Л о ж н ы й г е р м а ф р о д и т и з м, и н д у ц и р о в а н н ы й л е к а р с т в е н н ы м и п р е п а р а т а м и. В предыдущие годы андрогенизация нередко выявлялась у новорождённых девочек, матери которых принимали гестагенные или андрогенные препараты в 1-ом триместре беременности для предотвращения спонтанных абортов, а в более поздние сроки - для сохранения беременности при привычных выкидышах. В последние годы частота таких вариантов гермафродитизма снижается по мере прекращения использования подобных препаратов во время беременности.
В зависимости от времени использования препаратов, изменения в строении наружных половых органов напоминают те, которые отмечаются при врождённой дисфункции коры надпочечников, но в отличие от неё дальнейшего прогрессирования после рождения не отмечается.
Л о ж н ы й г е р м а ф р о д и т и з м в с л е д с т в и е г и п е р а н д р о г е н и и у м а т е р и и. Источником повышенной продукции андрогенов в организме беременной могут быть гормонально активные опухоли яичников (арренобластома, опухоль Крукенберга, лютеома, липоидноклеточная опухоль, опухоль из стромальных клеток яичника), реже надпочечников. У самой женщины гиперандрогения сопровождается гипертрофией клитора, акне, огрубением голоса, гипогалактией, избыточным оволосением. У новорождённых имеется тенденция к низкой массе тела при рождении и различной степени выраженности андрогенизация наружных половых органов.
И д и о п а т и ч е с к и й л о ж н ы й г е р м а ф р о д и т и з м. Выделяют две формы этого варианта гермафродитизма. Первая форма представляет собой небольшую по частоте группу новорождённых девочек, у которых андрогенизация наружных половых органов ассоциируется с врождёнными пороками развития ЖКТ и мочевыводящих путей (атрезия ануса, агенезия почек, обструктивные деформации мочевых путей, уретро-вагинальная фистула, дефекты формирования мюллеровых протоков). Андрогенизация наружных половых органов негормонального происхождения.
Вторая форма характеризуется только различной степенью андрогенизации наружных половых органов без сочетанных аномалий и пороков развития и отсутствием в анамнезе данных за какую-либо гиперандрогению. Не исключено, что эта форма может быть обусловлена неизвестными на сегодня нарушениями (ферментопатиями) в биосинтезе стероидов у плода или матери во время беременности, включая патологию плаценты.
Ложный мужской гермафродитизм. Этот тип гермафродитизма является следствием неполной маскулинизации плода мужского пола, причинами которой являются: дефекты биосинтеза тестостерона, дефекты рецепторов тестостерона (андроген-резистентные синдромы), а также тератогенное повреждение гонад или закладки гениталий (описаны выше).
В р о ж д ё н н а я д и с ф у н к ц и я к о р ы н а д п о ч е ч н и к о в (врождённая гиперплазия надпочечников). Классические варианты болезни у плода женского пола приводят к развитию ложного женского гермафродитизма (см. выше). У плода мужского пола в том случае, если дефект ферментов отмечается не только в надпочечниках, но и в тестикулах, то в последних нарушается биосинтез тестостерона, что нарушает формирование наружных половых органов по мужскому типу различной степени выраженности.
Подобный дефект 3b-оксистероиддегидрогеназы приводит к развитию тяжёлой сольтеряющей формы болезни, а степень изменений в строении наружных половых органов колеблется от коронковой формы гипоспадии до перинеосокральной формы, синдрома микропениса. Яички обычно располагаются в мошонке. В более редких случаях наследуются дефекты ферментов 17a-гидроксилазы, 17-кетостероидредуктазы и 17,20-лиазы. В двух последних вариантах отмечается строение наружных половых органов женского типа при отсутствии каких-либо структур внутренних половых органов.
А н д р о г е н - р е з и с т е н т н ы е с и н д р о м ы. Снижение или отсутствие чувствительности тканей к андрогенам развивается вследствие: дефекта фермента 5a-редуктазы, превращающей тестостерон в дигидротестостерон (ДГТ), а также как результат повреждения рецепторов тестостерона или пострецепторного механизма передачи его действия.
При дефекте 5a-редуктазы тестикулы продуцируют как тестостерон, так и МИФ, что обеспечивает регрессию мюллеровых структур и развитие вольфовых структур, как и в норме. Наружные же половые органы развиваются по мужскому типу только при достаточном уровне ДГТ, который при этом синдроме отсутствует. В результате наружные половые органы формируются по женскому типу: влагалище заканчивается слепо, половой член маленький и искривлённый, с закрытой крайней плотью и значительной гипоспадией. В периоде полового созревания на фоне повышения чувствительности тканей к самому тестостерону происходит маскулинизация (развитие мышечной системы и телосложения по мужскому типу), развитие лобкового оволосения, увеличение размеров полового члена и опущение яичек. При обследовании набор половых хромосом 46XY, значительно повышено соотношение тестостерон/ДГТ (>35), ещё более возрастающее на фоне стимуляции хорионическим гонадотропином (>74).
При дефекте рецепторов андрогенов (синдром тестикулярной феминизации) с полной нечувствительностью к ним набор половых хромосом 46XY молекулярными исследованиями выявлена мутация гена, контролирующего связывание андрогенов с рецепторами. Наследуется данный дефект сцепленно с Х-хромосомой. Уровень тестостерона после полового созревания соответствует мужским нормам. Наружные половые органы женского типа, нормально сформированные тестикулы могут располагаться в брюшной полости, в паховых каналах или больших половых губах, напоминая паховые «грыжы». Наружные 2/3 влагалища нормально развиты, заканчиваются слепо. В отличие от пациентов с дефектом 5a-редуктазы, в периоде полового созревания при этом синдроме не происходит маскулинизации, а грудные железы могут развиться вследствие периферического превращения резко повышенного тестостерона в эстрадиол. Отмечается достаточная эстрогенизация малых половых губ и влагалища. У большинства больных развивается небольшое лобковое оволосение, но примерно у 30% оволосение на лобке и в подмышечных впадинах отсутствует. Что касается роста, телосложения, тембра голоса пациенты достаточно фемининны. Они часто выходят замуж и не имеют проблем в половой жизни, но в некоторых случаях возникают трудности вследствие короткого влагалища и может потребоваться пластическая коррекция. До сих пор встречаются случаи поздней диагностики данного синдрома, когда пациенты впервые обращаются к врачу с жалобами на отсутствие менструаций или по поводу бесплодия, после операции грыжесечения и обнаружения тестикулярной ткани.
Встречаются случаи синдрома неполной тестикулярной феминизации, при которых чувствительность к андрогенам частично сохранена, включая синдром Райфенштейна (Raifenstein), при котором качественный дефект рецепторов или недостаточное связывание андрогенов с рецепторами нарушает андрогенизацию плода мужского пола, а в последующем способствует феминизации в периоде полового созревания. При наборе половых хромосом 46XY наружные половые органы или женского типа, или с различной степенью гермафродитизма.
Диагностические и дифференциально-диагностические критерии посиндромной оценки нарушения половой дифференцировки у новорождённых.
Несоответствие хромосомного пола и фенотипа новорождённого. Если при нормально сформированных по мужскому или женскому типу наружных половых органах (генитальный пол) их строение не совпадает с набором половых хромосом (генетический пол), то возможно несколько вариантов врождённых болезней и синдромов (табл. 1).
Таблица 1.
Болезни и синдромы, при которых генитальный пол не соответствует генетическому полу*
|
Название |
Генотип |
Фенотип |
Этиология |
|
Чистый гонадальный дисгенез 46ХХ Мужчины 46XY Женщины Врождённая липоидная гиперплазия Дефект 17,20-лиазы Дефект 17-гидрокси-лазы Андрогенрезистентные синдромы |
XY ХХ XY XY XY XY XY |
Женский Мужской Женский Женский Женский Женский Женский |
АР, ХСР SRY транслокация SRY делеция ВДКН ВДКН ВДКН ХСР/АД |
Примечание: АД – аутосомно-доминантный тип наследования; АР – аутосомно-рецессивный тип наследования; ВДКН – врождённая дисфункция коры надпочечников; SRY – ген, детерминирующий развитие тестикулярной ткани; ХСР – сцепленный с Х хромосомой рецессивный тип наследования; ХСР/АД – возможен как Х-сцепленный, так аутосомно-доминантный тип наследования.
Гипоспадия и крипторхизм. Около 25% новорождённых «мальчиков» с неопустившимися яичками и гипоспадией имеют те или иные нарушения половой дифференцировки. Частота изолированной гипоспадии составляет 1:125 новорождённых мальчиков и не связана с какими-либо генетическими или эндокринными нарушениями. В случаях с выраженной гипоспадией, сочетающихся с крипторхизмом, необходимо исключать ВДКН и другие варианты гермафродитизма.
Синдром микропениса. Изолированные варианты, обычно не связанные с нарушениями половой дифференцировки, тем не менее, требуют уточнения причин их развития. Этот синдром может отмечаться и при церебрально-гипофизарной недостаточности (см. ниже).
Неправильное или гермафродитное строение наружных половых органов. При неправильном строении гениталий у новорождённых, даже незначительно выраженном, необходимо как можно быстрее уточнить причину и определить пол ребёнка, в котором он в дальнейшем будет адаптирован. При поздней диагностике и задержке с принятием этого решения возможны серьёзные психологические и социальные проблемы у родителей, а у ребёнка тяжёлая психологическая травма и нарушения психо-сексуальной ориентации в более старшем возрасте. Родители должны быть немедленно информированы о том, что пол ребёнка не определён и на это может потребоваться несколько дней, чтобы провести все необходимые цитогенетические исследования и принять вместе с ними наиболее правильное решение. При разговоре с родителями не стоит слишком детально обсуждать ожидаемые результаты, особенно при несовпадении генетического и гонадного пола. Кроме того, при ВДКН часто необходимо проведение неотложной заместительной терапии, т.к. на 2-3-й день после рождения возможно развитие острой надпочечниковой недостаточности.
Неправильное строение гениталий является одним из проявлений гетерогенной группы болезней и синдромов, перечисленных в табл. 2.
Таблица 2.
Болезни и синдромы, сопровождающиеся нарушениями формирования наружных половых органов плода.
|
Андрогенизация плода женского пола Врождённая дисфункция коры надпочечников Дефицит 21-гидроксилазы Дефицит 11-гидроксилазы Дефицит 3b-оксистероид дегидрогеназы Аномалии расхождения половых хромосом X0/XY X/XY Варианты 2-х первых, повреждения одной из хромосом Избыток андрогенов в организме матери Приём андрогенов Повышенная продукция андрогенов в организме матери Истинный гермафродитизм Идиопатические формы Изолированные Сочетающиеся с врождёнными пороками развития срединных структур Гиперандрогенизация плода мужского пола Врождённая дисфункция коры надпочечников Дефицит 3b-оксистероид дегидрогеназы Андроген-резистентные синдромы Дефицит 5a-редуктазы Парциальный дефект рецепторов андрогенов Дисгенезия тестикул |
Обследование начинают с тщательно собранного анамнеза и клинического осмотра, после чего проводят необходимые лабораторные исследования.
При сборе анамнеза необходимо обратить внимание на следующие моменты во время беременности: использование лекарственных препаратов, особенно в 1-ом триместре беременности; признаки гиперандрогении (избыточное оволосение, огрубение голоса, акне, гипертрофии клитора); инфекционные болезни или воздействие неблагоприятных тератогенных факторов в 1-ом триместре беременности (могут вызвать парциальную дисгенезию гонад). Важными являются также данные о смерти сиблингов в первые 10 дней после рождения, наличие у них признаков андрогенизации или преждевременного полового созревания.
При осмотре новорождённого обращают внимание на следующие симптомы: наличие или отсутствие гонад при пальпации, длину и диаметр полового члена, микропенис или его агенезия при кариотипе 46XY, место открытия уретры, наличие или отсутствие влагалища, степень сращения больших половых губ, степень нарушения формирования мочевыводящих путей и анального отверстия.
На основании данных, полученных при сборе анамнеза и клиническом осмотре новорождённого, определяется необходимый объём лабораторных исследований, как неотложных, так и плановых (табл. 3).
Таблица 3.
Исследования, необходимые при выявлении отклонений в строении гениталий у новорождённых.*
|
Неотложные исследования Исследование кариотипа УЗИ малого таза (строение внутренних половых органов) Определение в сыворотке крови 17-оксипрогестерона 17-оксипрегненолола Тестостерона 11-дезоксикортизола Дигидротестостерона Плановые исследования Вагинография Диагностическая лапароскопия и лапаротомия, биопсия ткани гонад Рентгено-контрастные исследования ЖКТ и мочевыводящих путей Исследование активности фермента 5a-редуктазы в фибробластах |
*Для уточнения диагноза могут потребоваться дополнительные цитогенетические и гормональные исследования.
Необходимо отметить, что метод исследования кариотипа в костном мозге, несмотря на меньшее время исследования, менее точен по сравнению со стандартным его определением (не выявляет мозаичные формы) и может служить дополнительным, а не основным критерием. Только набор половых хромосом позволяет определить причину нарушения формирования гениталий - уточнить генетический пол ребёнка и сделать заключение о том, что эти изменения произошли или вследствие андрогенизации плода женского пола, или вследствие недостаточной андрогенизации плода мужского пола. Тем не менее, кариотип не всегда является основным критерием для выбора наиболее оптимального пола для данного ребёнка. Он окончательно определяется исходя из строения наружных гениталий с точки зрения их соответствия выбранному полу (с учётом их возможной пластической коррекции), а также обеспечения в дальнейшем нормальной психо-сексуальной адаптации данного индивидуума.
Ультразвуковое исследование строения внутренних половых органов новорождённого должно проводиться опытным специалистом для уточнения наличия или отсутствия матки и гонад. Обнаружение матки говорит о том, что функционирующая тестикулярная ткань отсутствует и, независимо от генетического пола, новорождённого лучше адаптировать в женском поле. Наоборот, отсутствие матки и других мюллеровых структур подтверждает наличие тестикулярной ткани, по крайней мере, на 7-9-й неделях беременности, и присутствия SRY гена или XY кариотипа. Но, как указывалось выше, это не может быть основанием для установления у новорождённого мужского пола, так как критериями для этого являются размеры полового члена и степень гипоспадии, а также уровень в крови тестостерона. Ультразвуковым методом могут быть также обнаружены не выявленные при пальпации гонады и определена их возможная половая принадлежность. Диагностические алгоритмы, основанные на исходных данных УЗИ о строении внутренних половых органов, представлены на рис. 2 и 3 и описывают этапы проведения всех необходимых исследований. В алгоритмы не включены пациенты с несоответствием генотипа и фенотипа. При этом диагноз не всегда является решающим фактором, определяющим решение о половой адаптации данного новорождённого.
Рис. 2. Алгоритм диагностики заболеваний и синдромов, вызывающих неправильное строение наружных половых органов у новорождённых со строением внутренних половых органов женского типа.
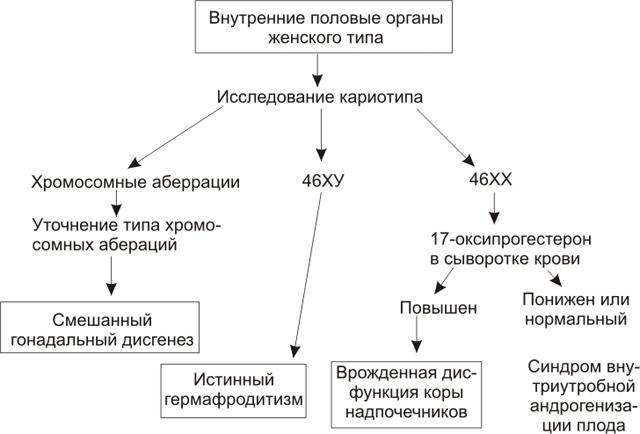
Рис. 3. Алгоритм диагностики заболеваний и синдромов, вызывающих неправильное строение наружных половых органов у новорождённых со строением внутренних половых органов мужского типа.
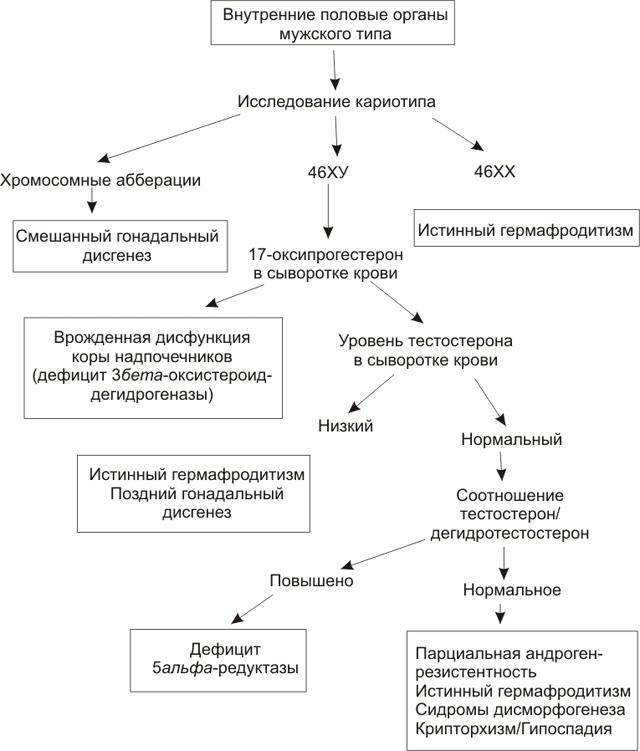
Важное значение для диагностики и решения вопроса о половой адаптации имеют определение в крови новорождённого различных стероидов (табл. 3), а также МИФ, гена SRY, кариотипа. На основании полученных данных принимается окончательное решение об адаптации ребёнка в том или ином паспортном поле с участием эндокринолога, уролога, гинеколога, иногда и психолога. После достижения согласованного решения проводится беседа с родителями ребёнка с полным и детальным его разъяснением, включающим прогноз дальнейшего полового развития и созревания, психо-сексуальной ориентации, фертильности, необходимости той или иной пластики наружных половых органов и заместительной гормональной терапии. В случаях синдрома неполной резистентности к андрогенам, смешанного гонадального дисгенеза и истинного гермафродитизма выбранный специалистами пол может не соответствовать истинному генетическому или гонадному полу новорожденного, т.к. он выбирается во многом исходя из особенностей строения наружных половых органов. Если у новорождённого половой член менее 4 см, имеет грубые аномалии развития, то вряд ли он сможет нормально адаптироваться в мужском паспортном поле.