

3 курс 1 часть / 0079436_7441E_eremin_v_v_kargov_s_i_uspenskaya
.pdfГ л а в а 1. Основы химической термодинамики |
67 |
4-13. Запишите выражение для расчета абсолютной энтропии одного моля воды при температуре 300 °С и давлении 2 атм.
4-14. Нарисуйте график зависимости стандартной энтропии воды от температуры в интервале от 0 до 400 К.
4-15. Запишите энтропию одного моля идеального газа как функцию температуры и давления (теплоемкость считать постоянной).
4-16. Определите зависимость энтропии от объема для термодинамической системы, которая описывается уравнением состояния (для одного моля):
|
p + |
a |
|
|
(V − b) = RT. |
|
|
|
|
||
TV |
2 |
||||
|
|
|
|
|
4-17. Определите зависимость энтропии от объема для термодинамической системы, которая описывается уравнением состояния (для одного моля):
|
|
|
|
b |
|
|
c |
|
|
pV = RT |
1 |
+ |
|
|
+ |
|
|
|
. |
V |
V |
2 |
|||||||
|
|
|
|
|
|
||||
4-18. Один моль газа описывается уравнением состояния:
( p + f (V ))(V − b) = RT ,
где f(V) – некоторая функция, которая не зависит от температуры. Рассчитайте изменение энтропии газа при его необратимом изотермическом расширении от объема V1 до объема V2.
4-19. Рассчитайте изменение энтропии 1000 г метанола в результате его замерзания при –105 °С. Теплота плавления твердого метанола при –98 °С (т.пл.) равна 3160 Дж моль–1. Теплоемкости твердого и жидкого метанола равны 55.6 и 81.6 Дж К–1 моль–1, соответственно. Объясните, почему энтропия при замерзании уменьшается, хотя процесс – самопроизвольный.
4-20. Теплоемкость некоторого вещества в интервале температур от T1 до T2 изменяется следующим образом:
C p
1 |
T 2 |
T 1 |
Постройте график зависимости энтропии вещества от температуры в этом интервале температур.
4-21. Пользуясь справочными данными, приведите пример самопроизвольной химической реакции, для которой стандартное изменение энтропии меньше 0.

68 |
Г л а в а 1. Основы химической термодинамики |
4-22. Пользуясь справочными данными, рассчитайте стандартное изме-
нение энтропии в реакции H2(г) + 1 O2(г) = H2O(г)
2
а) при 25 °С;
б) при 300 °С.
4-23. Рассчитайте изменение внутренней энергии, энтальпии и энтропии при нагревании 200 г воды от 25 °С до нормальной температуры кипения и полном испарении жидкости (давление нормальное). Примите, что мольная теплоемкость воды не зависит от температуры и равна: Cp = 75.3 Дж К–1 моль–1. Удельная теплота испарения воды при постоянном давлении равна 2260 Дж г–1.
4-24. Рассчитайте изменение внутренней энергии, энтальпии и энтропии при нагревании 200 г бензола от 25 °С до нормальной температуры кипения (80.1 °С) и полном испарении жидкости (давление нормальное). Примите, что мольная теплоемкость бензола не зависит от температуры и равна: Cp = 136.1 Дж К–1 моль–1. Удельная теплота испарения бензола при постоянном давлении равна 395 Дж г–1.
4-25. 3.00 моль газообразного CO2 расширяются изотермически (в тепловом контакте с окружающей средой, имеющей температуру 15.0 °C) против постоянного внешнего давления 1.00 бар. Начальный и конечный объемы газа равны 10.0 л и 30.0 л, соответственно. Рассчитайте изменение энтропии:
а) системы, считая CO2 идеальным газом; б) окружающей среды;
в) Вселенной (система плюс окружающая среда).
4-26. Стандартная энтропия золота при 25 °С: S298o = 47.40 Дж моль–1 К–1. При нагревании до 484 °С энтропия золота увеличивается в 1.5 раза. До какой температуры надо охладить золото, чтобы его стандартная энтропия была в два раза меньше, чем при 298 К? Теплоемкость можно считать не зависящей от температуры.
4-27. Стандартная энтропия алмаза при 25 °С: S298o = 2.38 Дж моль–1 К–1. При нагревании до 167 °С энтропия алмаза увеличивается вдвое. До какой температуры надо нагреть алмаз, чтобы его стандартная энтропия была в три раза больше, чем при 298 К? Теплоемкость можно считать не зависящей от температуры.
4-28. В ходе некоторого процесса система получила 1.50 кДж теплоты при 350 K. При этом энтропия системы изменилась на +5.51 Дж K–1. Можно ли считать этот процесс термодинамически обратимым? Ответ обоснуйте.
4-29. Докажите, что температурная шкала идеального газа и термодинамическая шкала температур, базирующаяся на втором законе термодинамики, совпадают с точностью до постоянного множителя.

Г л а в а 1. Основы химической термодинамики |
69 |
4-30. Покажите, что процесс смешения двух жидкостей с температурами T1 и T2 в изолированном сосуде является необратимым.
4-31. Можно ли поставить знак равенства между изоэнтропийным и адиабатическим процессом?
§ 5. Термодинамические потенциалы
Внутренняя энергия и энтропия относятся к классу характеристических функций. Функция называется характеристической, если все термодинамические свойства гомогенной системы могут быть выражены непосредственно через нее и ее частные производные по соответствующим переменным1. Эти независимые переменные называют естественными. Характеристические функции, по определению, содержат в себе всю термодинамическую информацию о системе. Но не все они одинаково удобны для решения конкретных задач. Так, некоторые из естественных переменных, например, энтропию, нельзя измерять (контролировать) в ходе какого-то процесса. Поэтому встает задача перехода от одних переменных к другим – экспериментально измеримым, но с условием сохранения характеристичности самой функции2. Такой переход осуществля-
ют с помощью преобразований Лежандра (см. пример 5-1).
С помощью этих преобразований вводятся другие характеристические функции:
•энтальпия
H(S, p, n) = U + pV,
•энергия Гельмгольца
F(T, V, n) = U – TS,
•энергия Гиббса
G(T, p, n) = U + pV – TS = H – TS = F + pV.
В скобках указаны их естественные переменные. Функции U, H, F, G
называют также термодинамическими потенциалами. Все потенциалы не имеют известного абсолютного значения, поскольку определены с точностью до постоянной, которая равна внутренней энергии при абсолютном нуле. Все они имеют размерность энергии.
Зависимость термодинамических потенциалов от их естественных переменных описывается основными уравнениями термодинамики –
1Понятие «характеристичность» связано с определенным набором переменных. Функция, характеристическая для одного набора переменных, перестает быть таковой при другом наборе.
2Сохранение характеристичности означает, что из данной характеристической функции с помощью некоторых преобразований мы получаем другую, из которой исходная функция восстанавливается однозначно, т.е. при этом в ней сохраняется вся исходная термодинамическая информация о системе.
70 |
Г л а в а 1. Основы химической термодинамики |
фундаментальными уравнениями Гиббса. В дифференциальной форме эти уравнения имеют вид:
(5.1.а)
(5.1.б)
(5.1.в)
(5.1.г)
(5.2)
(5.3)
|
∂U |
|
|
|
∂U |
|
dV + ∑ |
|
∂U |
|
dni = |
|
||||||||
dU = |
|
|
|
dS + |
|
|
|
|
|
|
|
|
|
|||||||
|
|
∂S V ,n |
|
|
∂V S,n |
|
i |
|
∂ni V ,S,n |
|
||||||||||
|
|
|
|
|
= TdS – pdV + ∑µ i dni |
|
|
|
|
|
i ≠ j |
|
||||||||
|
|
|
|
|
, |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
∂H |
|
i |
|
|
|
∂H |
|
|
|
|
|||
|
|
∂H |
|
|
|
|
|
|
dni = |
|
||||||||||
dH = |
|
dS + |
|
|
dp + ∑ |
|
|
|
|
|
|
|||||||||
|
|
∂S p,n |
|
|
∂p S,n |
|
i |
|
∂ni p,S,n |
|
||||||||||
|
|
|
|
|
= TdS + Vdp + ∑µ i dni |
|
|
|
|
|
i ≠ j |
|
||||||||
|
|
|
|
|
, |
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
i |
|
|
|
∂F |
|
|
|
|
|||
|
|
∂F |
|
|
|
|
∂F |
|
dV + ∑ |
|
|
|
|
|||||||
dF = |
|
|
|
dT + |
|
|
|
|
|
|
|
|
dni = |
|
||||||
|
|
|
|
|
|
|
||||||||||||||
|
|
∂T V ,n |
|
|
∂V T ,n |
|
i |
|
∂ni V ,T ,n |
|
||||||||||
|
|
|
|
= – SdT – pdV + ∑µ i dni , |
|
|
|
|
i ≠ j |
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
∂G |
|
i |
|
|
|
∂G |
|
|
|
|
|||
|
|
∂G |
|
|
|
dp + ∑ |
|
|
|
|
||||||||||
dG = |
|
|
dT + |
|
|
|
|
|
|
|
|
dni = |
|
|||||||
|
|
|
|
|
|
|
||||||||||||||
|
|
∂T p,n |
|
|
∂p T ,n |
|
i |
|
∂ni p,T ,n |
|
||||||||||
|
|
|
|
= –SdT + Vdp + ∑µ i dni |
|
|
|
|
|
i≠ j |
|
|||||||||
|
|
|
|
, |
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
i |
|
|
|
|
|
|
|
|
|
|
|
где химический потенциал |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
∂U |
|
|
|
∂H |
|
|
|
∂F |
|
|
|
|
|
|
∂G |
|
|||
µ i = |
|
|
= |
∂ni |
|
|
= |
|
|
|
|
|
|
|
= |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
∂ni S,V ,n |
i |
≠ j |
|
S, p,n |
i ≠ j |
|
∂ni T ,V ,n |
i≠ j |
|
∂ni T , p,n |
i≠ j |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
характеризует приращение соответствующего термодинамического потенциала при изменении количества данного вещества и постоянстве естественных переменных и количеств остальных веществ. Равенство химических потенциалов веществ является признаком их химического равновесия; разность химических потенциалов является движущей силой при массопереносе.
Если в системе совершается несколько видов работ, связанных с переносом вещества, то для описания равновесных состояний вместо химического потенциала используют понятие «полный потенциал», который помимо химического включает дополнительные вклады от этих видов работ. Например, при изменении электрического заряда системы на dq ее внутренняя энергия изменяется на ϕdq, где ϕ – электрический потенциал. Но заряд нельзя перемещать без материального носителя, поэтому, используя условие сохранения заряда q = F∑zi ni (F – число
i
Фарадея), уравнение (5.1.а) следует переписать в виде:
dU = TdS − pdV + ϕdq + ∑µ i dni = TdS − pdV + ∑µ i dni + ϕF ∑zi dni =
i |
i |
i |
= TdS − pdV + ϕdq + ∑(µ i + zi Fϕ)dni . |
|
|
i

|
|
|
|
|
Г л а в а 1. Основы химической термодинамики |
|
|
|
71 |
|||||||||||
|
|
|
В этом случае полный (электрохимический) потенциал i-го вещест- |
|
|
|||||||||||||||
ва |
|
i |
1 имеет вид: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
µ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
∂U |
|
|
∂H |
|
|
∂F |
|
|
∂G |
|
(5.4) |
|
||
|
|
|
|
|
|
|
|
|
|
|||||||||||
µ i = µ i + zi Fϕ = |
|
|
= |
|
|
= |
|
|
|
= |
|
|
|
|
||||||
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
∂ni S,V ,n |
i ≠ j |
|
∂ni S, p,n |
i ≠ j |
|
∂ni T ,V ,n |
i ≠ j |
|
∂ni T , p,n |
i ≠ j |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Аналогично, если некоторая фаза находится в поле тяготения и имеет большую протяженность вдоль градиента поля, то полный потенциал i-го компонента этой фазы записывается в виде:
µi* = µi + Migh, |
(5.5) |
где Mi – молярная масса i-го вещества, g – гравитационная постоянная, h – высота.
Зная любой из потенциалов как функцию естественных переменных, можно с помощью основного уравнения термодинамики найти все другие термодинамические функции и параметры системы. Для этого используют соотношения Максвелла. Рассмотрим, например, выражение (5.1.а) для внутренней энергии. Так как dU – полный дифференциал, частные производные внутренней энергии по естественным переменным равны:
|
∂U |
= T , |
|
∂U |
= − p . |
(5.6) |
|
|
|
|
|||
|
∂S V |
|
|
∂V S |
|
|
Если продифференцировать первое тождество по объему, а второе – по энтропии, то получатся смешанные вторые производные внутренней энергии, которые равны друг другу:
|
∂T |
|
∂p |
(5.7.а) |
||
|
|
|
= − |
|
. |
|
|
|
|||||
|
∂V S |
|
∂S V |
|
||
При перекрестном дифференцировании остальных уравнений (5.1) получаются еще три соотношения:
|
∂T |
|
∂V |
, |
(5.7.б) |
||||
|
|
|
= |
|
|
|
|||
|
∂p S |
|
∂S p |
|
|
||||
|
∂S |
|
∂p |
|
|
(5.7.в) |
|||
|
|
|
= |
|
|
|
|
, |
|
|
|
|
|
||||||
|
∂V T |
|
∂T V |
|
|
||||
∂V |
= − |
|
∂S |
|
|
(5.7.г) |
|||
|
|
|
∂p |
. |
|||||
∂T p |
|
|
T |
|
|||||
1 В настоящее время в электрохимии принято обозначать электрохимический потенциал чертой сверху, i . Следует обратить внимание, что это никак не связано с понятием парциального свойства, которое будет введено в § 6.
72 |
Г л а в а 1. Основы химической термодинамики |
Аналогичные равенства можно записать и для любых других термодинамических переменных, если воспользоваться условиями равенства смешанных производных функций состояния.
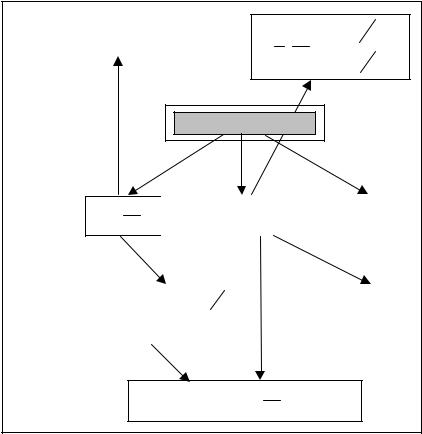
Свойство характеристичности термодинамической функции проиллюстрировано на рис. 5.1, где показано, как с помощью энергии Гиббса и ее частных производных по p,T могут быть выражены остальные термодинамические свойства системы.
Другой важный смысл термодинамических потенциалов состоит в том, что с их помощью можно указывать направление термодинамических процессов. Так, например, если процесс происходит при постоянных температуре и давлении, то неравенство (4.2), выражающее второй закон термодинамики, эквивалентно неравенству dGp,T ≤ 0 (см. пример 5-2), где знак равенства относится к обратимым процессам, а неравенства – к необратимым. Поэтому при необратимых процессах, протекающих при постоянных температуре и давлении, энергия Гиббса всегда уменьшается. Минимум энергии Гиббса соответствует равновесному состоянию системы.
|
|
∂S |
|
∂2 G |
|||
C p |
= T |
|
|
= −T |
|
|
|
|
∂T |
2 |
|||||
|
|
∂T p,n |
|
|
p,n |
||
α = 1 ∂V
V ∂T
|
= |
(∂2 G ∂T ∂p) |
|||
p,n |
(∂G |
∂p |
) |
n |
|
|
|
|
T ,n |
||
dG = −SdT + Vdp + ∑µi dni
S= − ∂G
∂T
|
|
|
|
∂G |
|
|
|
∂G |
|
|
|
V |
= |
|
|
µi |
= |
|
|
|
|
|
|||||||
p,n |
|
|
|
∂p T ,n |
|
|
|
∂ni p,T ,ni ≠ j |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
∂G |
|
|
|
∂ (G |
) |
|
|
∂G |
|||
|
2 |
|
T |
|
|
|
F = G − pV = G − p |
|
|
|||
|
|
|
||||||||||
H = G + TS = G −T |
|
|
= −T |
|
|
|
|
|
|
|
∂p T ,n |
|
|
|
|
|
|||||||||
|
∂T p,n |
|
|
|
∂T |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
p,n |
|
|
|
|
U= G + TS − pV = G −T ∂G
∂T
|
|
∂G |
|
− p |
|
p,n |
|
∂p T ,n |
Рис. 5.1 |
Связь между характеристической функцией – энергией Гиббса – |
|
и другими термодинамическими функциями |
|

Г л а в а 1. Основы химической термодинамики |
73 |
Аналогично, любой термодинамический потенциал в необратимых процессах при постоянстве естественных переменных уменьшается и достигает минимума при равновесии:
Потенциал |
Естественные |
Условие само- |
Условия |
|
переменные |
произвольности |
равновесия |
|
|
|
|
U |
S = const, V = const |
dU < 0 |
dU = 0, d 2U > 0 |
H |
S = const, p = const |
dH < 0 |
dH = 0, d 2H > 0 |
F |
T = const, V = const |
dF < 0 |
dF = 0, d 2F > 0 |
|
|
|
dG = 0, d 2G > 0 |
G |
T = const, p = const |
dG < 0 |
Наибольшее значение в практических термодинамических расчетах имеют два последние потенциала – энергия Гельмгольца F и энергия Гиббса G, т.к. их естественные переменные наиболее удобны для измерения и фиксирования в ходе химических превращений. Функции F и G имеют дополнительный физико-химический смысл. Изменение энергии Гельмгольца в каком-либо процессе при T = const, V = const равно максимальной работе, которую может совершить система при обратимом процессе:
F1 – F2 = –Wmax. |
(5.8.а) |
Таким образом, энергия F равна той части внутренней энергии (U = F + TS), которая может превратиться в работу.
Аналогично, изменение энергии Гиббса в каком-либо процессе при T = const, p = const равно максимальной полезной (т.е. немеханической) работе, которую может совершить система в этом процессе:
G1 – G2 = –Wпол. |
(5.8.б) |
Расчет изменения энергии Гиббса и энергии Гельмгольца в различных процессах
1. Изменение энергий Гиббса и Гельмгольца при изменении температуры
Зависимость энергии Гиббса и Гельмгольца от температуры в закрытых системах может быть определена с помощью фундаментальных уравнений:
dF = −SdT − pdV , dG = −SdT + Vdp
или с помощью уравнения Гиббса – Гельмгольца:
|
∂(F /T ) |
= − |
U |
|
|
∂(G /T ) |
= − |
H |
(5.9) |
|||
|
|
|
|
, |
|
|
|
|
. |
|||
T |
2 |
T |
2 |
|||||||||
|
∂T V |
|
|
|
|
∂T p |
|
|
|
|
||

74 |
Г л а в а 1. Основы химической термодинамики |
(5.10.а)
(5.10.б)
Функции F и G являются функциями состояния, для них:
|
∂F |
= −SV , |
|
|
|
|
∂T V |
|
|
∂G |
= −Sp . |
|
|
|
|
∂T p |
|
Для интегрирования этих уравнений надо знать температурную зависимость энтропии, которая определяется теплоемкостью системы (формулы (4.21.а)). График зависимости энергии Гиббса1 от температуры в предположении линейной температурной зависимости теплоемкости приведен на рис. 5.2.а.
|
(а) |
G–H0 |
G–H0 |
|
T |
(б)
газ
конд. фаза
p
Рис. 5.2
(5.11.а)
(5.11.б)
Зависимость энергии Гиббса:
(a) от температуры при p = const, (б) от давления при T = const
2. Изменение энергий Гиббса и Гельмгольца при изотермическом расширении или сжатии
Зависимость энергии Гиббса и Гельмгольца от давления и объема при постоянной температуре может быть определена интегрированием
производных: |
|
|
∂F |
= − p , |
|
|
|
|
∂V T |
|
|
|
∂G |
= V . |
|
|
|
|
∂p T |
|
1 Энергия Гиббса вещества отсчитана относительно условно выбранного уровня H0.

Г л а в а 1. Основы химической термодинамики |
75 |
Для восстановления вида этой зависимости необходимо знать урав- нение состояния фазы. Так, для идеального газа
V |
nRT |
||
F(V2 ) − F(V1) = − ∫2 |
|||
V |
|||
V |
|||
1 |
|
|
|
p |
nRT |
||
G( p2 ) − G( p1) = ∫2 |
|
||
|
p |
||
p1 |
|||
dV = nRT ln V1 , V2
dp = nRT ln p2 . p1
Если p1 = p° = 1 бар, то говорят о стандартной энергии Гиббса, G°
G( p) − G o ( p o ) = nRT ln p .
На рис. 5.2.б приведены графики зависимости энергии Гиббса от давления при T = const для конденсированной фазы и газа.
3. Изменение энергий Гиббса и Гельмгольца при химической реакции
Расчет изменения функций F и G в химических реакциях можно проводить разными способами. Рассмотрим три из них на примере энергии Гиббса.
I. По определению, G = H – TS. Если продукты реакции и исходные вещества находятся при одинаковой температуре, то изменение энергии Гиббса в химической реакции
∑νi Ai = ∑ν j B j i j
(5.12.а)
(5.12.б)
(5.12.в)
равно
∆rGT = ∑ν jGj (T ) − ∑νiGi (T ) = ∆r HT − T ∆r ST . |
(5.13.а) |
|
j |
i |
|
В стандартных условиях |
|
|
∆rGTo = ∑ν jGoj |
(T ) − ∑νiGio (T ) = ∆r HTo − T ∆r STo . |
(5.13.б) |
j |
i |
|
II. Аналогично тепловому эффекту реакции, изменение энергии Гиббса можно рассчитать, используя стандартные энергии Гиббса образования реагентов и продуктов:
∆rGTo = ∑ν j ∆f Goj |
(T ) − ∑νi ∆f Gio (T ) |
|
(5.14) |
||
j |
|
i |
|
|
|
или |
|
|
|
|
|
∆rGTo = ∑ν j ∆f H oj |
|
|
− |
∑νi Sio |
|
− ∑νi ∆f Hio − T ∑ν j Soj |
. |
||||
j |
i |
j |
|
i |
|
Для расчета ∆rG при условиях, отличных от стандартных, используют соотношение (5.11.б).

76 |
|
Г л а в а 1. Основы химической термодинамики |
|
|
Изменение энергии Гельмгольца химической реакции между иде- |
|
|
альными газами связано с энергией Гиббса |
(5.15) |
∆rG = ∆r F + ∆νRT , |
|
|
|
где ∆ν – изменение количества молей газообразных веществ в ходе ре- |
|
|
акции. Для реакций в конденсированной фазе при небольших давлениях |
(5.16) |
∆rG ∆r F . |
|
III. Стандартная энергия Гиббса реакции может быть рассчитана с помощью стандартных приведенных потенциалов:
(5.17.а)
(5.17.б)
(5.18.а)
(5.18.б)
Фo(T ) = − Go(T ) − H o(0) ,
T
Ф′o(T ) = − Go(T ) − H o(298) ,
T
где G°(T) – стандартное значение энергии Гиббса при температуре Т, H°(0), H°(298) – стандартные значения энтальпии при температурах 0 и 298 К.
Функции Ф°(T) и Ф′°(T) вычисляются для газов по молекулярным данным, а для конденсированных фаз – на основании экспериментальных данных по теплоемкости. Связь между приведенными потенциалами и стандарной энергией Гиббса реакции выражается соотношениями:
|
|
|
|
|
|
|
+ ∆ r H o (0) |
|
∆ rG o (T ) = −T ∆ rФo (T ) + ∆ r H o (0) = −T ∑ν jФoj |
−∑ν iФio |
, |
||||||
|
|
|
j |
|
i |
|
|
|
|
|
|
|
|
|
|
|
|
∆ rG o (T ) = −T ∆ rФ′o (T ) + ∆ r H o (298) = |
|
|
|
|||||
|
|
|
|
+ ∆ r H o (298) |
, |
|
|
|
= −T |
∑ν jФ′jo −∑ν iФi′o |
|
|
|
||||
|
j |
i |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
где ∆rH°(0), ∆rH°(298) – стандартные энтальпии реакции при 0 и 298 К.
ПРИМЕРЫ
Пример 5-1. Выполните преобразования Лежандра для функций: а) одной переменной y = cos(x),
б) двух переменных U = U (S,V).
Решение. Преобразования Лежандра являются примером так называемых контактных преобразований и имеют вид:
Х= x′ (х), Y(x) = xy′(x) – y(x), Y′(X) = x,
х= Y′(Х), y(x) = ХY ′(X) – Y(X), y′(x) = X.