Патогенез острого ишемического и реперфузионного повреждения миокарда
Активное клиническое применение тромболитических препаратов и методов эндоваскулярных вмешательств при остром коронарном синдроме позволило снизить смертность от инфаркта миокарда, но привело к осознанию того, что восстановление кровотока в ишемизированном миокарде вызывает дополнительное повреждение сердечной мышцы. Реперфузионное повреждение миокарда требует понимания его механизмов и внедрения в клиническую практику новых способов защиты .
Патогенез ишемического повреждения миокарда
Чаще всего в основе развития ишемического повреждения миокарда лежит нарушение притока артериальной крови по одной коронарных артерий, возникающее чаще всего в результате тромбоза артерии в месте разрыва атеросклеротической бляшки.
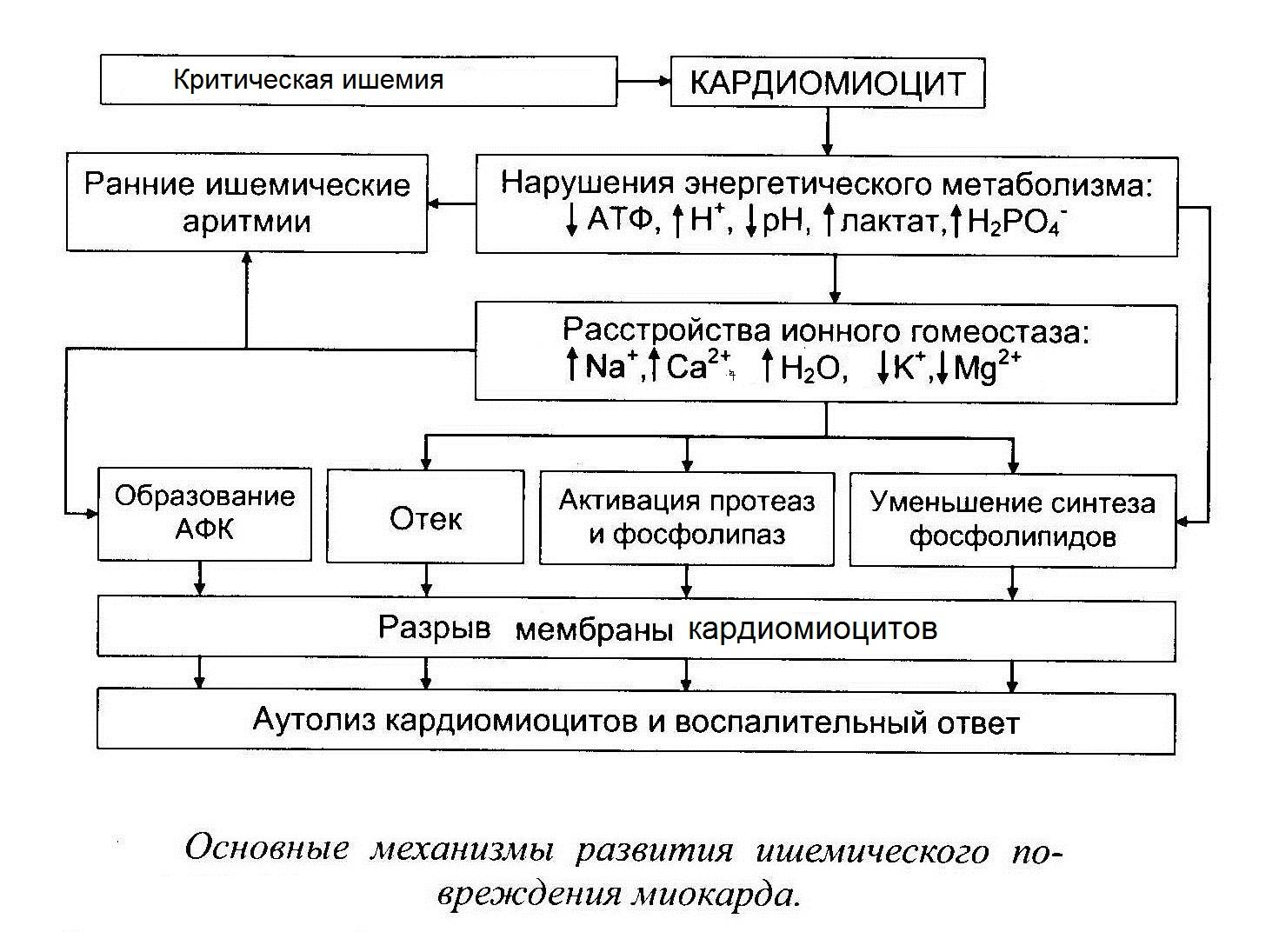
При ишемии наиболее ранние изменения возникают в системе энергетического метаболизма кардиомиоцитов. Так, транспорт электронов по дыхательной цепи митохондрий и образование АТФ путем окислительного фосфорилирования прекращаются в течение нескольких секунд после наступления полной ишемии. Практически одновременно происходит активация ключевых ферментов гликогенолиза (фосфорилазы) и гликолиза (фосфофруктокиназы). Дополнительная активация фосфорилазы осуществляется за счет высвобождения катехоламинов из ишемизированных нервных окончаний и стимуляции β-адренорецепторов кардиомиоцитов с последующим возрастанием внутриклеточной концентрации циклического аденозинмонофосфата. В итоге гликоген включается в гликолитический путь.
Запасы высокоэнергетических фосфатов, среди которых важнейшую роль играет АТФ, в миокарде невелики. При острой ишемии потребность в АТФ многократно превышает скорость ее образования, и единственным ее источником при этом является гликолиз. Гликолитическая продукция АТФ также быстро снижается вследствие накопления в миокарде конечных продуктов гликолиза - протонов, лактата и НАДН.
Отсутствие кислорода и торможение гликолиза являются не единственными причинами снижения уровня АТФ в ишемизированном миокарде. Снижению АТФ при ишемии также способствует нарушение ее транспорта из митохондрий и активация некоторых циклических метаболических процессов, в ходе которых происходит усиленная утилизация АТФ. К таким процессам относится избыточный захват кальция (Са2+) в саркоплазматический ретикулум (СР) и его высвобождение из СР, а также распад триглицеридов до свободных жирных кислот с последующим их ресинтезом. Эти процессы усугубляют и без того выраженный энергетический дефицит клеток миокарда.
Метаболические нарушения в ишемизированном миокарде затрагивают также обмен липидов и белков. При ишемии средней тяжести, в частности, при наличии коллатерального кровотока, происходит усиление захвата жирных кислот (ЖК) в зоне ишемии. Скорость захвата ЖК при этом превышает скорость их утилизации, что приводит к накоплению избытка свободных ЖК, ацил-КоА и ацилкарнитина, которые в высоких концентрациях обладают выраженным повреждающим действием в отношении функций сарколеммы и митохондрий.
При тяжелой ишемии происходит полное угнетение митохондриального окисления ЖК и единственным источником энергии служит анаэробный метаболизм глюкозы. Наступление лактат-ацидоза и гибель клетки при этом происходят значительно быстрее.
Важнейшим следствием нарушений энергетического метаболизма миокарда в ранней стадии ишемии является его сократительная дисфункция, возникающая в результате:
Истощения запасов высокоэнергетических фосфатов, в первую очередь, АТФ и креатинфосфата.
Накопления конечных продуктов метаболизма, в частности, протонов (Н+), лактата, неорганического фосфата и пуриновых оснований, которые создают «осмотическую нагрузку».
Развития внутриклеточного ацидоза, ухудшающего сократительную функцию вследствие вытеснения Са2+ из актомиозиновых комплексов.
Резкого сниженя перфузионного давления в дистальных сосудах ишемизированного отдела сердца, что изменяет биомеханические свойства стенки ЛЖ.
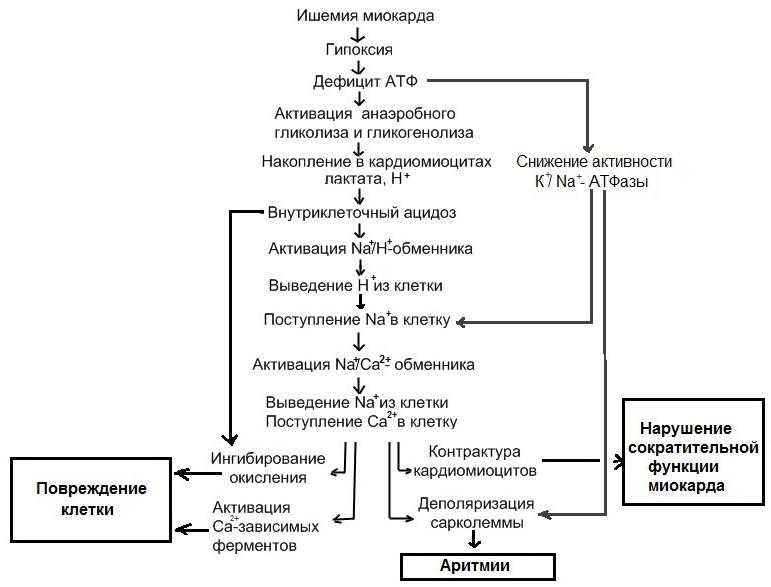
В раннем периоде ишемии происходит возрастание внутриклеточной концентрации Са2+, вследствие его поступления по потенциал-зависимым Са2+ каналам, выхода его из СР, а также возрастания его притока в результате активации «обратного» варианта работы Na+/Ca2+обменника (выведение Na+ из клетки в обмен на поступающий внутрь нее Са2+). Данный вариант работы Na+/Ca2+обменника активируется в ходе ишемии вследствие повышения внутриклеточной концентрации Na+, поступающего в клетку в обмен на протоны, выводимые Na+/H+ насосом для компенсации внутриклеточного ацидоза. Повышение внутриклеточной концентрации Са2+ является ключевым звеном патогенетической цепи, ведущей к необратимому повреждению кардиомиоцитов, т.к. приводит активации Са2+-зависимых протеаз, липаз и фосфолипаз, ингибированию биологического окисления в митохондриях и их повреждению, деполяризации сарколеммы и ишемической контрактуре кардиомиоцитов.
Критическим событием в патогенезе ишемического повреждения миокарда является переход обратимой стадии повреждения в необратимую. Последняя характеризуется гибелью кардиомиоцитов в результате некроза и апоптоза.
Важнейшим механизмом необратимого повреждения кардиомиоцитов является нарушение целостности их мембраны посредством следующих механизмов:
Активация перекисного окисления липидов.
Усиленное образование амфифильных липидов, к которым относятся свободные ЖК, ацил-КоА, ацилкарнитин и лизофосфолипиды, нарушающих активность интегральных мембранных белков - ферментов, рецепторов и ионных каналов.
Активация лизосомальных ферментов.
Активация протеаз которые разрушают цитоскелета кардиомиоцитов.
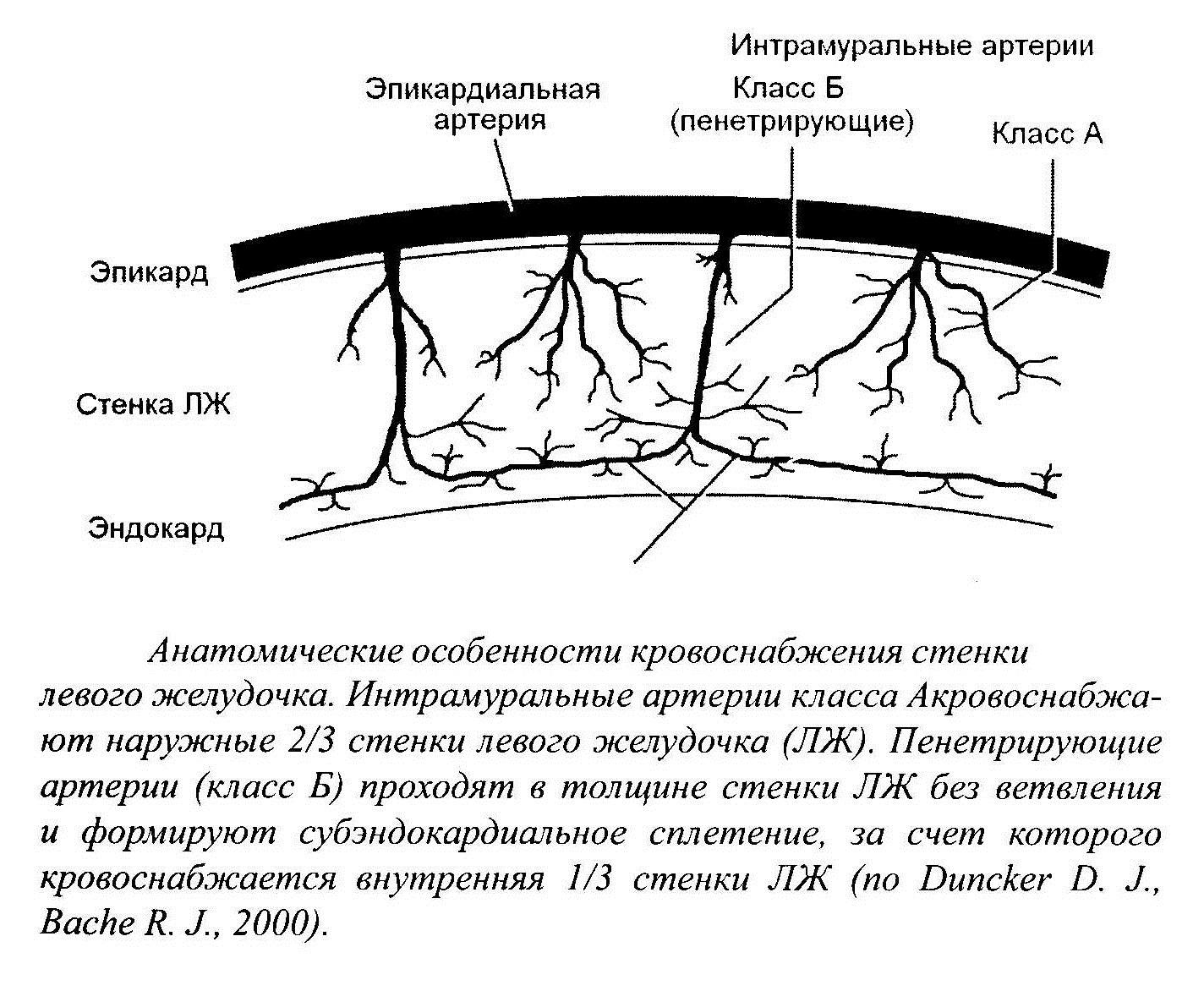
Распространение зоны некроза в ишемизированном миокарде происходит в виде «волны клеточной гибели», зарождающейся в субэндокарде и постепенно, с течением времени, распространяющейся по направлению к эпикардиалъной поверхности. Это объясняется тем, что внутренние слои миокарда наиболее удалены от проходящих субэпикардиально коронарных артерий и, кроме того, подвергаются наибольшему механическому сдавлению в систолу.