

MedGen розширена
.PDFI
1 |
2 |
3 |
4 |
5 |
II |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
1 |
|
|
|
2 |
3 |
|
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
5 |
|
|
|
6 |
7 |
8 |
|
|
|
|
|
9 |
|
|
||||||||||||||||||||
III |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
2 |
|
3 |
4 |
5 |
|
|
6 |
|
7 |
|
|
|
|
8 |
|
|
9 |
|
|
10 |
|
|
11 |
|
|
|||||||||||||||||||||
IV |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
Рис. 4.8. Родовід з двома жінками, гомозиготними за гемофілією. Батьки жінок — двою- |
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||||||||||
|
|
рідні брат і сестра (за Ф. Фогель і А. Мотульски) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
P &ХАХа |
|
|
× |
|
|
|
%ХаY |
|
|
I |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
G |
|
ХА, Ха |
|
|
|
|
|
|
|
Ха, Y |
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
F1 |
|
ХАХа, ХАY, |
|
ХаХа, |
|
|
ХаY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
хвора |
хворий |
здорова |
|
здоровий |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
дівчинка хлопчик |
дівчинка |
|
хлопчик |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
5. Від хворого батька ознаку успадковують усі |
|
II |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
дочки і ніколи — сини, оскільки син успадковує |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
7 |
||||||||||||||||||||
|
|
|
|
|
1 |
|
|
|
2 |
|
|
|
3 |
|
|
4 |
|
5 |
|
6 |
|
||||||||||||||||||||||||||||||||||
від батька Y-хромосому. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
P &ХаХа |
× |
|
|
|
%ХАY |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
G |
|
|
Ха |
|
|
|
|
|
|
ХА, Y |
|
|
III |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
F1 |
|
|
|
|
|
|
ХАХа, |
|
|
ХаY |
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
2 |
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
4 |
|
|
|
|
|
5 |
|||||||||
|
|
|
|
|
|
|
|
хвора |
|
здоровий |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
дівчинка |
|
хлопчик |
|
|
|
Рис. 4.9. Родовід при Х-зчепленому домінант- |
|||||||||||||||||||||||||||||||||||||||||||
6. У жінок захворювання спостерігається в |
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ному типі успадкування |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||
легшій формі, оскільки вони частіше гетерозиготні |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
(ХАХа). У гемізиготних чоловіків (ХАY) захворю- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
вання виявляється у важчій формі. Домінантний |
|
батька до сина, родоводи характеризуються на- |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
ген може давати летальний ефект у ембріона чо- |
|
ступним (рис. 4.11): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||
ловічої статі. У такому разі всі хворі в родоводі |
|
|
1. Хворіють тільки чоловіки. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||||||||
тільки жіночої статі (рис. 4.10). |
|
|
|
|
|
|
|
2. Від хворого батька ознаку успадковують усі |
|||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
сини. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Y-зчеплений тип успадкування |
|
|
|
Патологічні мутації, що стосуються спермато- |
|||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
генезу, успадковуватися не можуть, оскільки хворі |
|||||||||||||||||||||||||||||||||||||||||||||||||||||
У Y-хромосомі знаходяться гени, які контролю- |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
стерильні. Але можлива передача гена при екст- |
||||||||||||||||||||||||||||||||||||||||||||||||||||||
ють сперматогенез, ріст тіла, кінцівок і зубів, ово- |
|
ракорпоральному заплідненні. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||
лосіння вушної раковини та ін. (голандричні оз- |
|
|
Всі розглянуті вище типи успадкування пов’я- |
||||||||||||||||||||||||||||||||||||||||||||||||||||
наки). Оскільки Y-хромосома передається від |
|
зані з мутаціями ядерної ДНК. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||||||||||||
I |
|
|
|
1 |
2 |
3 |
|
4 |
|
|
|
|
Рис. 4.10. Родовід при |
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Х-зчепленому типі успадку- |
|
|
|
|
|
|
|
|
|
|
|
|
вання у разі летального ефек- |
|
|
|
|
|
|
|
|
|
|
|
|
ту патологічного гена в емб- |
|
|
|
|
|
|
|
|
|
|
|
|
ріона чоловічої статі (синд- II |
|
|
|
|
|
|
|
|
|
|
|
|
ром Блоха — Сульцбергера) |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
69

I |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
КОНТРОЛЬНІ ПИТАННЯ |
||||||
|
|
|
|
|
|
1 |
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
ДО РОЗДІЛУ 4 |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1. Що означають терміни генеалогія, пробанд, |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
сибси? |
||||||
II |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2. Значення клініко-генеалогічного методу. |
||||||
1 |
|
|
|
|
2 |
|
3 |
|
|
|
|
4 |
|
|
5 |
|
|
|
6 |
|
|
3. Особливості збору генеалогічного анамнезу. |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4. Генеалогічна символіка. Правила графічно- |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
го зображення родоводу. |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5. Характерні особливості родоводу при авто- |
|||||
III |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
сомно-домінантному, автосомно-рецесивному і |
|||||||
1 |
|
|
|
|
|
2 |
|
|
|
|
|
3 |
|
|
|
4 |
|
|
5 |
|
зчепленому зі статтю типах успадкування. |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6. Особливості родоводу при мітохондріально- |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
му успадкуванні. |
||||||
Рис. 4.11. Родовід при Y-зчепленому успадку- |
|
|
|
|
|
|
|
||||||||||||||||||||||||||
ванні ознаки (оволосіння вушної раковини) |
|
|
|
|
Контрольно-навчальні питання |
||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Завдання 1 |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Оберіть одну відповідь. |
|||||
4.2.4. РОДОВОДИ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1. Сибси — це: |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
А. Батьки пробанда |
||||||||||||||||
ПРИ МІТОХОНДРІАЛЬНОМУ |
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
В. Діти пробанда |
|||||||||||||||||||||||||
УСПАДКУВАННІ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
С. Брати і сестри пробанда |
|||||||||||||||
Мітохондріальні хвороби, що пов’язані з мута- |
|
D. Родичі пробанда, особисто обстежені ліка- |
|||||||||||||||||||||||||||||||
рем-генетиком |
|||||||||||||||||||||||||||||||||
ціями мітохондріальної ДНК, характеризуються |
|
|
|
|
|
|
|
||||||||||||||||||||||||||
мітохондріальним або цитоплазматичним типами |
|
2. Батьки хворої дитини здорові, але аналогічні |
|||||||||||||||||||||||||||||||
успадкування. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
захворювання зустрічаються у сибсів хворого (не- |
|||||||||||
Родоводи при мітохондріальному успадкуванні |
залежно від статі). Це найбільш характерно для |
||||||||||||||||||||||||||||||||
мають такі особливості: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
такого типу успадкування: |
||||||||||||||||
1. Хвороба передається від хворої матері всім |
|
А. Автосомно-домінантного |
|||||||||||||||||||||||||||||||
її дітям, як синам, так і дочкам (рис. 4.12). |
|
|
|
|
В. Автосомно-рецесивного |
||||||||||||||||||||||||||||
2. Передача хвороби по чоловічій лінії немож- |
|
С. Рецесивного, зчепленого з Х-хромосомою |
|||||||||||||||||||||||||||||||
лива. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D. Домінантного, зчепленого з Х-хромосомою |
||||||
При мультифакторіальних хворобах, розвиток |
|
Е. Мітохондріального |
|||||||||||||||||||||||||||||||
яких пов’язаний з кількома генами схильності і |
|
|
|
|
|
|
|
||||||||||||||||||||||||||
дією факторів середовища, родоводи характеризу- |
|
3. Для родоводу з автосомно-домінантним ти- |
|||||||||||||||||||||||||||||||
ються сімейним накопиченням випадків хвороби, |
пом успадкування характерно: |
||||||||||||||||||||||||||||||||
але не відповідають жодному з перерахованих |
|
А. Ознака успадковується «за вертикаллю»: у |
|||||||||||||||||||||||||||||||
типів успадкування (див. рис. 7.2). |
|
|
|
|
|
|
|
хворої дитини, як правило, хворий один із батьків |
|||||||||||||||||||||||||
I |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
2 |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
II
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
III
1 |
2 |
3 |
4 |
5 |
|
6 |
7 |
8 |
9 |
|
|
|
|
1 |
2 |
3 |
|
4 |
5 |
|
Рис. 4.12. Родовід при мітохондріальному успадкуванні ознаки |
|
|
|
|||||
70

В. Хворіють жінки і чоловіки однаково часто, |
|
I |
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
ризик народження хворої дитини у гетерозиготних |
|
|
|
|
1 |
|
|
|
|
|
2 |
|
|
3 |
||||||
батьків 25 % |
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
С. Від хворого батька ознаку успадковують |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
100 % дочок і ніколи — сини |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
D. Хворіють переважно чоловіки; якщо мати |
|
II |
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
гетерозиготна, то 50 % синів можуть бути хвори- |
|
1 |
|
|
2 |
|
|
3 |
|
|
4 |
|
||||||||
ми |
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
Е. Хворіють переважно чоловіки; від хворого |
|
Рис. 4.13. Родовід сім’ї з трьома випадками |
||||||||||||||||||
батька хворобу успадковують 100 % синів |
|
|||||||||||||||||||
ахондроплазії в одному поколінні (у здорового |
||||||||||||||||||||
|
|
|
|
|||||||||||||||||
4. Для родоводу з автосомно-рецесивним типом |
чоловіка від першого шлюбу із здоровою жінкою |
|||||||||||||||||||
двоє дочок з ахондроплазією — II,1 і II,3; і від дру- |
||||||||||||||||||||
успадкування характерно: |
гого шлюбу із здоровою жінкою — дочка з ахон- |
|||||||||||||||||||
А. Ознака успадковується «за вертикаллю»: у |
||||||||||||||||||||
дроплазією — II,4) |
|
|
|
|
|
|
|
|
|
|||||||||||
хворої дитини, як правило, хворий один із батьків |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
В. Хворіють жінки і чоловіки однаково часто, |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
ризик народження хворої дитини у гетерозиготних |
|
С. Рецесивний, зчеплений з Х-хромосомою |
||||||||||||||||||
батьків 25 % |
|
D. Домінантний, зчеплений з Х-хромосомою |
||||||||||||||||||
С. Від хворого батька ознаку успадковують |
|
Е. Мітохондріальний |
|
|
|
|
|
|||||||||||||
100 % дочок і ніколи — сини |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
D. Хворіють переважно чоловіки; якщо мати |
|
8. На рисунку (рис. 4.13) подано родовід сім’ї з |
||||||||||||||||||
гетерозиготна, то 50 % синів можуть бути хвори- |
3 дітьми з ахондроплазією у здорового чоловіка |
|||||||||||||||||||
ми |
від двох шлюбів зі здоровими жінками. Ахондро- |
|||||||||||||||||||
Е. Хворіють переважно чоловіки, від хворого |
плазія — автосомно-домінантне захворювання. |
|||||||||||||||||||
батька хворобу успадковують 100 % синів |
Пенетрантність гена 100 %. Найвірогідніше пояс- |
|||||||||||||||||||
5. При якому типі успадкування хворіють пе- |
нення народження трьох дітей з ахондроплазією |
|||||||||||||||||||
в цій сім’ї: |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
реважно чоловіки? |
|
А. Гонадний мозаїцизм у батька |
|
|
||||||||||||||||
А. Автосомно-домінантному |
|
В. Випадкові генеративні мутації гена у батька |
||||||||||||||||||
В. Автосомно-рецесивному |
|
С. Результат соматичних мутацій у дітей в про- |
||||||||||||||||||
С. Рецесивному, зчепленому з Х-хромосомою |
цесі ембріонального розвитку |
|
|
|||||||||||||||||
D. Домінантному, зчепленому з Х-хромосомою |
|
D. Уніпарентна дисомія — успадкування двох |
||||||||||||||||||
Е. Мітохондріальному |
гомологічних хромосом від батька |
|
|
|||||||||||||||||
6. При якому типі успадкування хворіють час- |
|
Е. Варіювальна експресивність гена |
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
тіше жінки? |
|
9. На рисунку (рис. 4.14) подано родовід сім’ї, |
||||||||||||||||||
А. Автосомно-домінантному |
частина членів якої страждають на атрофію зоро- |
|||||||||||||||||||
В. Автосомно-рецесивному |
вого нерва типу Лебера. Для якого типу успадку- |
|||||||||||||||||||
С. Рецесивному, зчепленому з Х-хромосомою |
вання найбільш характерний такий родовід? |
|||||||||||||||||||
D. Домінантному, зчепленому з Х-хромосомою |
|
А. Автосомно-домінантного |
|
|
||||||||||||||||
Е. Мітохондріальному |
|
В. Автосомно-рецесивного |
|
|
||||||||||||||||
7. Інформація про походження подружжя і їхніх |
|
С. Рецесивного, зчепленого з Х-хромосомою |
||||||||||||||||||
|
D. Домінантного, зчепленого з Х-хромосомою |
|||||||||||||||||||
батьків з одного або близько розташованих |
|
Е. Мітохондріального |
|
|
|
|
|
|||||||||||||
пунктів може свідчити про такий тип успадкуван- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
ня хвороби: |
|
10. Якщо перша дитина у здорових батьків на- |
||||||||||||||||||
А. Автосомно-домінантний |
родилася з фенілкетонурією (автосомно-рецесив- |
|||||||||||||||||||
В. Автосомно-рецесивний |
на ознака), найвірогідніший висновок генетика: |
|||||||||||||||||||
I |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
II
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
III
1 |
2 |
3 |
4 |
5 |
6 |
7 |
Рис. 4.14. Родовід сім’ї, частина членів якої страждають на атрофію зорового нерва типу Лебера
71
А. Це результат соматичної мутації у хворої дитини
В. Це результат нової генеративної мутації в одного з батьків
С. Батьки — гетерозиготні носії рецесивного патологічного гена (Аа обидва)
D. Мати або батько — гетерозиготи за домінантним патологічним геном, який має неповну пенетрантність
Е. Це результат хромосомної мутації в одного з батьків
Завдання 2
Побудуйте родовід.
1. Пробанд — здорова жінка 23 років. Звернулася до медико-генетичної консультації за прогно-
зом потомства. У пробанда двоє здорових братів
ібрат та сестра, які страждають на алькаптонурію. Мати пробанда здорова і має здорових сестру і брата. Батько пробанда хворий на алькаптонурію
іє двоюрідним дядьком своєї дружини. У нього є здорові брат і сестра. Бабуся по лінії батька була хворою і перебувала в шлюбі з своїм здоровим двоюрідним братом. Бабуся і дідусь по лінії матері здорові, батько і мати діда також здорові, при цьому мати діда — рідна сестра діда пробанда з боку батька. Чоловік пробанда здоровий, 25 років. Його мати, батько, два брати здорові. У сім’ї не було хворих з алькаптонурією. Визначте тип успадкування алькаптонурії. Розрахуйте ризик народження хворих дітей у пробанда.
72

Розділ 5
ХРОМОСОМНІ ХВОРОБИ
5.1. ІСТОРІЯ ЦИТОГЕНЕТИКИ ЛЮДИНИ
Хромосомні хвороби — це спадкові хвороби, зумовлені зміною кількості і структури хромосом. Розділ генетики, який вивчає будову і функції хромосом різних організмів, називається цитогенетикою. Цитогенетика як наука розвивається з початку ХХ ст. У 1902 р. Сеттон і Бовері вперше висловили припущення про те, що гени знаходяться в хромосомах. У 1903 р. Сеттон запровадив термін «цитогенетика». У 1911 р. Томас Морган сформулював основні положення хромосомної теорії спадковості.
Майже всі важливі відкриття у галузі цитогенетики тварин і рослин були зроблені в першій половині ХХ ст. Цитогенетика людини почала реально розвиватися тільки в 50-ті роки XX ст. після того, як Тіо і Леван (J. Tjio, А. Levan) в 1956 р. ви-
користали нову методику для виготовлення препаратів хромосом людини і встановили, що в каріотипі людини 46 хромосом. У 1959 р. французький цитогенетик Лежен відкрив трисомію за 21-ю хромосомою при синдромі Дауна, а Форд і співробітники (1959), Джекобс і Стронг (1959) повідоми-
Таблиця 5.1. Хромосомні та геномні мутації
в матеріалах спонтанних абортів
|
Від загальної |
|
Тип мутації |
кількості абортованих |
|
ембріонів з хромосомними |
||
|
||
|
і геномними мутаціями, % |
|
|
|
|
Триплоїдія |
15 |
|
Тетраплоїдія |
5 |
|
Моносомія Х |
20 |
|
Трисомії: |
|
|
за 13-ю хромосомою |
2 |
|
за 16-ю хромосомою |
15 |
|
за 18-ю хромосомою |
3 |
|
за 21-ю хромосомою |
5 |
|
інші |
25 |
|
Інші мутації |
10 |
|
|
|
ли про каріотип 47,XXY при синдромі Клайнфельтера і каріотип 45,Х при синдромі Шерешевського — Тернера. У 1960 р. були відкриті причини синдромів Патау й Едвардса. У 1963 р. Лежен описав перший синдром, пов’язаний з хромосомною делецією, — синдром «котячого крику». У 1964 р. Джекобс припустив існування зв’язку між каріотипом 47,XYY і кримінальною психопатією. У 1968–1970 рр. були розроблені методи диференціального забарвлення хромосом, що дозволило однозначно ідентифікувати всі хромосоми людини і вивчити хромосомні аберації.
5.2. ЗНАЧЕННЯ ХРОМОСОМНИХ І ГЕНОМНИХ МУТАЦІЙ В ОНТОГЕНЕЗІ
Не менше 10 % сперматозоїдів і 25 % зрілих овоцитів мають хромосомні або геномні мутації. Від 35 до 50 % зародків людини гинуть на стадії бластоцисти, тобто до імплантації. У людини ця рання загибель ембріонів зазвичай не розпізнається. У значного відсотка з них спостерігаються хромосомні перебудови.
Приблизно 15 % усіх зареєстрованих вагітностей закінчується спонтанними абортами, близько половини яких обумовлено хромосомними аномаліями. У матеріалі спонтанного аборту часто знаходять поліплоїдію (20 %), моносомію Х (20 %), повні трисомії за автосомами (50 %), інші хромосомні і геномні мутації — в 10 % випадків (табл. 5.1). Трисомії за деякими автосомами (1, 5, 6, 11, 19) у елімінованих ембріонів і плодів зустрічаються вкрай рідко, що свідчить про велике значення цих хромосом для ембріонального розвитку. Ці аномалії порушують гаметогенез або переривають розвиток у доімплантаційному періоді.
Чим раніше переривається вагітність, тим більше вірогідно, що це спричинено хромосомними або геномними мутаціями. Так, якщо аборт відбувається в перші 2–4 тиж, то хромосомні аномалії спостерігаються у 60–70 % абортованих ембріонів. У першому триместрі вагітності хромосомні аномалії зустрічаються у 50 % абортусів, у
73

другому триместрі — у 30 %, у 20–27 тиж — у 7 % загиблих плодів, і, нарешті, 6 % мертвонароджень обумовлено хромосомною патологією.
Якщо порушення ембріонального розвитку сумісні з життям, то народжується дитина з хромосомною хворобою. Хромосомні хвороби виявляються у 0,5–1 % живонароджених, а у новонароджених із множинними вродженими вадами розвитку їх частота зростає до 40 %.
Хромосомні та геномні мутації мають значення не тільки в патології ранніх періодів онтогенезу (загибель гамет, незачаття, спонтанний аборт, мертвонародження, хромосомна хвороба). Хромосомні аномалії виникають у постнатальному періоді в 2 % соматичних клітин (соматичні мутації). Вони можуть бути причиною злоякісних пухлин. Наприклад, транслокація між хромосомами 9 і 22 (при цьому утворюється філадельфійська хромосома) є частою причиною хронічного мієлолейкозу.
В цілому хромосомні та геномні мутації можуть бути причиною таких клінічних ситуацій.
1. Безплідний шлюб. Від 2 до 4 % безплідних пар мають хромосомні й геномні мутації. Вони призводять до порушення сперматогенезу, овогенезу або до загибелі ембріонів до імплантації. Безплідність може бути наслідком:
—носійства збалансованих хромосомних му-
тацій;
—мікроделеції довгого плеча Y-хромосоми у чоловіків у ділянці, де знаходяться гени, що кодують сперматогенез, — фактор азооспермії (AZF);
—синдрому Клайнфельтера у чоловіків (каріо-
тип 47, ХХY);
—синдрому Шерешевського — Тернера у жінок (45,Х) та інших причин.
5.3. КЛАСИФІКАЦІЯ ХРОМОСОМНИХ ХВОРОБ
Як зазначалося вище, хромосомні хвороби — це спадкові хвороби, зумовлені зміною кількості і структури хромосом. Сьогодні описано понад 1000 таких хвороб. Близько 100 з них мають чітку клінічну картину і називаються синдромами. В основі класифікації хромосомних хвороб лежать три принципи (табл. 5.2).
Всі хромосомні хвороби можна розділити на три групи залежно від характеру зміни каріотипу (етіологічний принцип).
Хромосомні хвороби
Полі- |
Захворювання, |
Захворювання, |
плоїдії |
пов’язані зі зміною |
пов’язані зі змі- |
|
кількості |
ною кількості і |
|
і структури |
структури стате- |
|
автосом |
вих хромосом |
Залежно від типу клітин, в яких виникають мутації (і відповідно від відсотка клітин із зміненим каріотипом), розрізняють повні і мозаїчні форми хромосомних хвороб. Повні форми є наслідком генеративної мутації у батьків (тобто мутації відбуваються при утворенні статевих клітин у батьків). Усі клітини ембріона мають змінений каріотип. Мозаїчні форми є наслідком соматичної мутації, яка виникає у самого ембріона в процесі
2.Спонтанні аборти і мертвонародження. ембріонального періоду розвитку. Тому частина
3.Народження дитини з хромосомною хвороклітин хворого має нормальний набір хромосом,
бою, симптомами якої можуть бути множинні |
а решта — змінений. Мозаїчні форми, як прави- |
вроджені вади розвитку, відставання в психомо- |
ло, перебігають легше, ніж повні. Для виникнен- |
торному розвитку у дітей раннього віку, розумо- |
ня мозаїчної форми, яка за клінічною картиною |
ва відсталість у старших, відставання в рості, |
збігається з повною формою, необхідна наявність |
безплідність. |
не менше 10 % клітин з аномальним каріотипом. |
4. Злоякісні пухлини, особливо лейкоз. |
Проте слід зазначити, що немає повного парале- |
Таблиця 5.2. Класифікація хромосомних хвороб
Принцип класифікації |
Форми хромосомних хвороб |
|
|
За характером зміни каріотипу (етіологічний |
— поліплоїдії |
принцип, тобто характеристика |
— зміна кількості і структури автосом |
хромосомної або геномної мутації) |
— зміна кількості і структури статевих хромосом |
|
|
Залежно від типу клітин, в яких виникають |
— повні форми — результат генеративної мутації |
мутації |
у батьків |
|
— мозаїчні форми — результат соматичної мутації |
|
у самого ембріона |
|
|
Час виникнення мутації (у поколіннях) |
— спорадичні — результат нової мутації |
|
— успадковані — успадковуються від батьків |
|
зі збалансованими хромосомними мутаціями |
|
або від батьків з хромосомними хворобами |
|
|
74
лізму між кількістю уражених клітин і тяжкістю захворювання. Іноді при незначному відсотку аномальних клітин може бути тяжкий перебіг хвороби і навпаки.
Залежно від часу, коли виникла мутація в поколіннях, виділяють спорадичні хромосомні хвороби (наслідок нової мутації) і успадковані (успадковуються від батьків зі збалансованою мутацією або з хромосомною хворобою). Описано випадки народження дітей у хворих із синдромами Клайнфельтера, полісомії Х у жінок, полісомії Y у чоловіків, у жінок з синдромом Дауна. Чоловіки з синдромом Дауна, як правило, безплідні, оскільки у них порушується сперматогенез.
Таким чином, для точної діагностики хромосомної хвороби необхідно визначити:
1)тип мутації;
2)залучену в процес хромосому;
3)форму (повна або мозаїчна);
4)вид хвороби (спорадичний випадок або успадкована форма).
Така діагностика можлива тільки при цитогенетичному дослідженні, що проводиться у пацієнта, а іноді й у його батьків і сибсів.
5.4. ПАТОГЕНЕЗ ХРОМОСОМНИХ ХВОРОБ
Патогенез хромосомних хвороб дуже складний. Він залежить від порушення експресії великої кількості генів, залучених у хромосомну або геномну мутацію. Симптоми хромосомних хвороб обумовлені найчастіше так званим ефектом дози генів (мутації призводять або до збільшення, або до зменшення кількості генів), рідше відбувається порушення структури генів. Дія хромосомних і геномних мутацій починає виявлятися в ембріональному періоді розвитку. Найбільш ранні етапи дроблення зиготи контролюються речовинами, накопиченими в яйцеклітині. Потім відбувається включення власних генів зиготи. Загалом в ембріональному періоді працюють близько 1000 генів, що відповідають за різні етапи онтогенезу. При геномних і хромосомних мутаціях порушується баланс великої кількості генів, у тому числі й генів, що регулюють ембріональний розвиток. Спостерігаються також зміни експресії генів, пов’язані з геномним імпринтингом. Це неминуче призводить до порушення гістогенезу і органогенезу, тому хромосомні хвороби виявляються вродженими вадами розвитку.
Частіше порушення виявляються несумісними з життям, що призводить до внутрішньоутробної загибелі ембріона або плода. Рідше народжується дитина з хромосомною хворобою. Клінічно хромосомні хвороби виявляються синдромами множинних вроджених вад розвитку. Практично всі вони формуються до моменту народження. Винятком є зміни кількості і структури статевих хромосом. Остаточний фенотип у хворих із дисбалансом по статевих хромосомах може формуватися в підлітковому періоді.
Генетики порівнюють хромосомні хвороби з попелищем після пожежі. Пожежа — це те, що відбувається в ембріональному періоді. До моменту народження формується остаточний фенотип (головешки після пожежі). Нічого виправити вже неможливо. Можна лише провести косметичну корекцію, прооперувати хворого з вадою розвитку (якщо синдром сумісний з життям).
Оскільки при хромосомних хворобах порушуються ранні етапи ембріонального розвитку, то уражаються водночас багато органів і систем органів. Це робить клінічну картину багатьох хромосомних хвороб схожою. Чим більшим є дисбаланс хромосомного матеріалу, тим більш неспецифічна клініка хромосомної хвороби (особливо це стосується клінічної картини поліплоїдій). При мікроделеціях і мікродуплікаціях клінічна картина може бути дуже специфічною.
Для будь-якої хромосомної хвороби характерний поліморфізм, оскільки індивідуальний генотип особин впливає на експресію генів.
5.5. ЗАГАЛЬНІ СИМПТОМИ ХРОМОСОМНИХ ХВОРОБ
Хромосомні хвороби виявляються як синдроми множинних вроджених вад розвитку. Діагностичні ознаки хромосомних синдромів можна розділити на три групи:
1.Загальні ознаки, що дозволяють запідозрити аномалії хромосом (психічне або фізичне недорозвинення, черепно-лицьові дизморфії, зміни дерматогліфіки, вади внутрішніх органів).
2.Ознаки, що найчастіше зустрічаються при певних синдромах. Наприклад, при синдромі Едвардса в 90 % випадків зустрічається доліхоцефалія і в 96 % — флексорне згинання кисті. При синдромі Патау в середньому в 70 % випадків зустрічаються щілина губи і піднебіння, мікрофтальмія, полікістоз нирок, полідактилія. При синдромі Дауна більш як у 90 % випадків відзначається монголоїдний розріз очей і в 60 % — чотирипальцева поперечна складка на долоні.
3.Ознаки, патогномонічні для певного синдрому. Наприклад, при синдромі «котячого крику» відзначається характерний крик, що нагадує котяче нявкання. Як правило, поліплоїдії та зміни кількості і структури автосом виявляються певними симптомами в період вагітності та діагностуються у новонароджених. Хромосомні хвороби, пов’язані зі зміною кількості і структури статевих хромосом, відрізняються більш легким перебігом.
ЗАГАЛЬНІ СИМПТОМИ ПОЛІПЛОЇДІЇ І АНОМАЛІЙ АВТОСОМ
У період вагітності
1.Загроза переривання вагітності.
2.Затримка внутрішньоутробного розвитку (діагностується на підставі серії ультразвукових досліджень).
75
3. Маловоддя або багатоводдя. |
— фенотипічні порушення більшою мірою по- |
||
4. |
Токсикози у період вагітності. |
значаються на розвитку статевих органів і рості, |
|
5. Передчасні пологи. Рідше нормальний термін |
спостерігаються вади розвитку, особливо при син- |
||
вагітності. |
дромі Шерешевського — Тернера, але зустріча- |
||
Загальні симптоми |
ються вони рідше і менш тяжкі; |
||
— остаточний фенотип, як правило, формуєть- |
|||
1. |
Гіпотрофія або гіпоплазія при народженні. |
ся в підлітковому періоді; винятком є синдром |
|
Шерешевського — Тернера, при якому чіткі |
|||
2. |
Відставання в психомоторному розвитку у |
||
дітей раннього віку. |
клінічні ознаки виявляються у новонароджених; |
||
— можливі легкі форми, які виявляють тільки |
|||
3. |
Розумова відсталість у старших дітей (зазви- |
||
при популяційних дослідженнях. |
|||
чай тяжка). |
|
||
4.Затримка росту.
5.Безплідність.
Дизморфії обличчя і черепа
1.Мікроцефалія.
2.«Дизморфічне обличчя»: мікроаномалії очей (монголоїдний або антимонголоїдний розріз, гіпоабо гіпертелоризм, епікант та ін.), низько розташовані й деформовані вушні раковини; мікрогенія (гіпоплазія нижньої щелепи) та ін.
5.6. КЛІНІКО-ЦИТОГЕНЕТИЧНА ХАРАКТЕРИСТИКА НАЙПОШИРЕНІШИХ ХРОМОСОМНИХ ХВОРОБ І СИНДРОМІВ
Верхні та нижні кінцівки |
Клінічну і цитогенетичну характеристику ос- |
|
1. Чотирипальцева складка на долоні, єдина |
новних хромосомних хвороб і синдромів наведе- |
|
но в табл. 5.3. |
||
згинальна складка на 5-му пальці й інші зміни дер- |
|
|
матогліфіки, клинодактилія 5-го пальця, синдак- |
Поліплоїдія |
|
тилія, полідактилія. |
||
Триплоїдія. Мозаїчні форми триплоїдії описані |
||
2. Сандалеподібна щілина на стопі. |
||
3. Стопа-гойдалка. |
ще на початку 60-х років, а повна форма — впер- |
|
4. Поперечна згинальна борозна на підошві. |
ше в 1967 р. Частота в популяції 1:10 000 у ново- |
|
|
народжених. |
|
Внутрішні органи |
Каріотип при повній формі 69,ХХY або |
|
Часто зустрічаються вроджені вади серця і ве- |
69,XXX, при мозаїчній — 69,ХХХ/46,ХХ або інші |
|
(див. рис. 2.14). |
||
ликих судин, мозку, сечостатевої системи, травно- |
У більшості випадків триплоїдія — летальна |
|
го тракту та інших систем органів, порушення |
||
мутація, причому летальний ефект спостерігаєть- |
||
функції ендокринної та імунної систем. |
ся в ембріональному періоді. Триплоїди, що мають |
|
Як виняток зустрічаються такі симптомоком- |
||
два хромосомні набори матері й один хромосом- |
||
плекси: |
ний набір батька, абортуються в ранніх термінах |
|
1. Розумова відсталість без якихось вад розвит- |
||
вагітності. Триплоїди з двома хромосомними на- |
||
ку. |
борами батька більш життєздатні, оскільки у них |
|
2. Вади розвитку при нормальному психічно- |
||
краще розвинені позазародкові оболонки і плацен- |
||
му розвитку. |
та. Це часта причина міхурового заносу. Дуже |
|
3. Ізольовані (поодинокі) вади розвитку. |
||
рідко такі триплоїди завершують ембріональний |
||
|
||
|
розвиток (рис. 5.1). |
|
СИМПТОМИ ХВОРОБ, |
Спостерігається токсикоз другої половини ва- |
|
гітності з багатоводдям, збільшується вміст хоріо- |
||
ЗУМОВЛЕНИХ ЗМІНОЮ |
нічного гонадотропіну (ХГТ). Діти народжують- |
|
КІЛЬКОСТІ І СТРУКТУРИ |
ся недоношеними з гіпоплазією. Середня три- |
|
СТАТЕВИХ ХРОМОСОМ |
валість вагітності 36 тиж, маса при народженні, в |
|
|
середньому, 1300 г (максимум — 2000 г). |
|
Хромосомні хвороби, зумовлені зміною кіль- |
Діагноз триплоїдії можна встановити при на- |
|
кості і структури Х-статевих хромосом, як прави- |
родженні дитини. Характерні різке збільшення |
|
ло, характеризуються легшим перебігом. Легша |
маси і розмірів плаценти, осередкова псевдокістоз- |
|
клінічна картина цих синдромів обумовлена інак- |
на дегенерація ворсин хоріона. Середня тривалість |
|
тивацією зайвих Х-хромосом в ембріональному |
життя — 9 днів, більша тривалість життя відміче- |
|
періоді розвитку. |
на у мозаїків. |
|
Для цих хромосомних хвороб характерне таке: |
При поліплоїдії мало специфічних симптомів, |
|
— розумовий розвиток часто нижчий за нор- |
оскільки наявний дисбаланс по всьому хромосом- |
|
му, але аномалії розвитку мозку виражені не так |
ному набору. Як правило, у хворих дуже велике |
|
помітно, як при аномаліях автосом. Багато хворих |
заднє тім’ячко, низько розташовані вушні ракови- |
|
мають нормальний інтелект, а деякі — вище серед- |
ни з недорозвиненими мочками, вади очей (мікро- |
|
нього; |
фтальм, колобома райдужки, катаракта, дисплазія |
76
Таблиця 5.3. Основні хромосомні хвороби (частоти за С. І. Козловою і співавт., 1996, Н. П. Бочковим, 2001)
|
Синдром |
Каріотип |
Частота в популяції |
Основні клінічні симптоми |
|
у новонароджених |
|||
|
|
|
|
|
|
|
|
|
|
|
Триплоїдія |
69,ХХY або 69,XXX |
1:10 000 |
Збільшення маси і розмірів плаценти, осередкова псевдокістозна дегенерація |
|
|
|
|
ворсин хоріона. Гіпоплазія при народженні. Летальний синдром множинних |
|
|
|
|
вроджених вад розвитку |
|
|
|
|
|
|
Синдром Дауна |
47,ХХ,+21 |
1:700–1:800 |
Розумова відсталість, м’язова гіпотонія, брахіцефалія, мікроцефалія, плоске |
|
|
або 47,ХY,+21 |
|
обличчя, монголоїдний розріз очей, макроглосія, в 50 % вади серця, вади |
|
|
|
|
інших органів, імунодефіцитні стани. Деякі хворі живуть до 50–60 років |
|
|
|
|
|
|
Синдром Едвардса |
47,ХХ, +18 |
1:5000–1:7000 |
Пренатальна гіпоплазія, єдина пупкова артерія, доліхоцефалія, нависла |
|
|
або 47,ХY, +18 |
(70 % хворих |
потилиця, мікрогенія, специфічне згинання пальців кисті, стопа-гойдалка, |
|
|
|
— дівчатка) |
множинні вади розвитку внутрішніх органів, летальний синдром |
|
|
|
|
|
|
Синдром Патау |
47,ХХ, +13 |
1:5000–1:7000 |
Мікроцефалія, щілина губи і піднебіння, полідактилія, множинні вроджені |
|
|
або 47,ХY, +13 |
|
вади розвитку внутрішніх органів, летальний синдром |
|
|
|
|
|
|
Синдром |
46,ХХ, del5p- |
1:45 000–1:50 000 |
Незвичайний крик, що нагадує котяче нявкання, мікроцефалія, |
|
«крику кішки» |
або 46,XY, del 5p- |
|
антимонголоїдний розріз очей, гіпертелоризм, широке перенісся, вади |
|
|
|
|
внутрішніх органів, розумова відсталість. Деякі хворі живуть більше 50 років |
|
|
|
|
|
|
Синдром Ангельмана |
46,ХХ, del 15q- |
1:50 000 |
Мікробрахіцефалія, подовжене обличчя, макростомія, тяжка розумова |
|
(синдром |
або 46,XY, del 15q- |
|
відсталість, груба затримка мовного розвитку, судоми, характерна хода, що |
|
«щасливої ляльки») |
(мутація успадковується |
|
нагадує рухи механічної ляльки, легко провоковані або спонтанні напади сміху |
77 |
|
від матері) |
|
(звідси назва — синдром «щасливої ляльки»). Хворі часто висолоплюють язик |
|
|
|
|
|
|
Синдром Прадера — |
46,ХХ, del 15qабо |
1:25 000– |
М’язова гіпотонія, гіпогонадизм, ожиріння, розумова відсталість, маленькі |
|
Віллі |
46,XY, del 15q- (мутація |
1:50 000 |
кисті і стопи |
|
|
успадковується від батька) |
(ОМІМ) |
|
|
|
|
|
|
|
Синдром |
45,Х |
1:3000–1:3500 |
У новонароджених — лімфатичний набряк кистей і стіп, особливо добре |
|
Шерешевського — |
|
дівчаток |
помітний на нижніх кінцівках; гіпотонія, шкірні складки на шиї. |
|
Тернера |
|
|
У старших дітей — статевий інфантилізм, первинна аменорея, низький зріст, |
|
|
|
|
шкірні складки на шиї, вроджені вади серцево-судинної, сечостатевої |
|
|
|
|
та інших систем. Інтелект, як правило, нормальний |
|
|
|
|
|
|
Полісомії Х синдром |
Частіше трисомія Х — |
1:1000–1:1200 |
Клінічна картина трисомії Х варіабельна — від практично здорових |
|
(синдром «супержінка») |
47,ХХХ, рідко тетрасомія |
дівчаток |
фертильних жінок до пацієнток з вираженим гіпергонадотропним |
|
|
— 48,XXXХ і ще рідше |
|
гіпогонадизмом, безплідністю, олігофренією. При тетрасомії Х і пентасомії Х |
|
|
пентасомія 49,XXXXХ |
|
більш виражена симптоматика, в 100 % олігофренія. Описано черепно- |
|
|
|
|
лицьові дизморфії, вади зубів, скелета і статевих органів |
|
|
|
|
|
|
Синдром |
Частіше трисомія — |
1:1000 |
При трисоміях варіабельна клініка від легких форм з нормальним інтелектом |
|
Клайнфельтера |
47,XXY, рідко |
хлопчиків |
і фертильністю до тяжких, які супроводжуються гіпогеніталізмом, гіпогонадиз- |
|
|
тетрасомія — |
|
мом, безплідністю, олігофренією. При тетрасоміях і пентасоміях завжди вираже- |
|
|
48,XXXY або 48,XXYY |
|
на симптоматика. Зовнішні ознаки: євнухоїдна статура, подовжені дистальні |
|
|
і пентасомія — 49,XXXXY |
|
відділи кінцівок, гінекомастія, оволосіння за жіночим типом, гіпогонадизм |
|
|
|
|
|
|
Синдром полісомії |
Частіше трисомія |
1:1000 |
Клінічні симптоми варіюють від практично нормальних чоловіків |
|
Y-хромосоми (синдром |
47,ХYY, рідко |
хлопчиків |
за фізичним і розумовим розвитком до пацієнтів з легкою розумовою |
|
«суперчоловік») |
48,ХYYY, 49,ХYYYY |
|
відсталістю, схильністю до агресивних і навіть кримінальних вчинків |
|
|
|
|
|
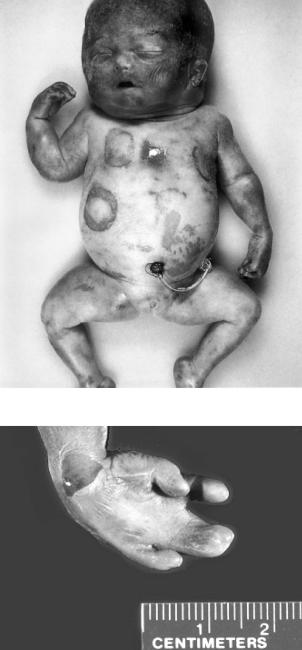
Рис. 5.1. Новонароджений із триплоїдією
Рис. 5.2. Синдактилія 3-го і 4-го пальців у плода з триплоїдією
сітківки, помутніння рогівки), гіпертелоризм, мікрогенія, мікростомія, щілина губи і піднебіння. Часто зустрічається синдактилія 3-го і 4-го пальців кисті (рис. 5.2), клино- і камптодактилія. На стопі синдактилія 3–5-го, нерідко 2-го і 3-го пальців.
Часто спостерігаються вади внутрішніх органів: ЦНС (спинномозкова грижа, прозенцефалія з гідроцефалією, циклопія, цебоцефалія та ін.), сечостатевої, серцево-судинної системи, шлунковокишкового тракту, залоз внутрішньої секреції, статевих органів (крипторхізм, епіабо гіпоспадія, гіпоплазія статевого члена та ін.).
Медико-генетичне консультування: повторні випадки народження сибсів з триплоїдією невідомі.
Тетраплоїдія. (92,ХХХХ) — відомі поодинокі спостереження, летальна мутація.
Синдром Дауна (трисомія 21)
Вперше в самостійну нозологічну одиницю цей синдром виділив англійський лікар Даун (Down) в 1866 р. під назвою «монголоїдна ідіотія». Етіологія захворювання встановлена набагато пізніше
— J. Lejeune і співавтори (1959) знайшли у таких хворих додаткову 21-шу хромосому.
Основні діагностичні ознаки: розумова відста-
лість, м’язова гіпотонія, брахіцефалія, мікроцефалія, плоске обличчя, монголоїдний розріз очей, макроглосія, вади серця та інших органів, імунодефіцитні стани, трисомія за 21-ю хромосомою.
Хвороба Дауна — найпоширеніша форма хромосомної патології людини. Частота в популяції у новонароджених 1:700–1:800. Співвідношення хлопчиків і дівчаток серед новонароджених із синдромом Дауна дорівнює 1:1.
У94 % усіх форм синдрому Дауна виявляється повна трисомія. Каріотип при повній трисомії
47,ХХ,+21 або 47,ХY,+21 (рис. 5.3). У 80 % ви-
падків зайва хромосома має материнське походження і лише в 20 % — батьківське. У 4 % випадків зустрічається робертсонівська транслокація (рис. 5.4) і в 2 % — мозаїцизм (табл. 5.4).
Основні діагностичні ознаки і клініка захворювання настільки типові й добре описані в літературі, що діагноз встановлюється вже в період новонародженості (рис. 5.5). За даними різних авторів, при хворобі Дауна зустрічається від 9 до 29 мікроаномалій і вад розвитку.
Діти з синдромом Дауна народжуються, як правило, в строк з помірною гіпоплазією. Маса тіла при народженні в середньому 2900 г. Найхарактернішими симптомами у новонароджених є м’язова гіпотонія і гіпорефлексія в поєднанні з розхитаністю суглобів. Брахіцефальний череп зі сплощеною потилицею. Обличчя кругле сплощене, монголоїдний розріз очей, епікант (рис. 5.6), плями Брушфільда (світлі плями на райдужці), розширене і сплощене перенісся, маленькі низько розташовані вушні раковини (мікротія) із закрученим завитком. Великий, зазвичай висолоплений, язик (макроглосія), високе піднебіння. Шия коротка, характерний надлишок шкіри на шиї. Кисті широкі, короткі, клинодактилія мізинців, одна згинальна борозна на мізинці (через гіпоплазію серединної фаланги), чотирипальцева складка на долоні (рис. 5.7). На стопі сандалеподібна щілина.
Перераховані симптоми зустрічаються не у 100 % хворих. У табл. 5.5 наведена частота зовнішніх ознак синдрому Дауна.
У50 % хворих зустрічаються вроджені вади серця (дефекти міжшлуночкової і міжпередсердної перегородок, відкрита артеріальна протока та ін.),
Таблиця 5.4. Цитогенетичні порушення
при синдромі Дауна
Мутації |
Частота, % |
|
|
Повна трисомія |
94 |
Транслокація |
4 |
Мозаїцизм |
2 |
|
|
78