

MedGen розширена
.PDF1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
Рис. 5.3. Каріотип чоловіка з |
|
|
|
|
|
|
синдромом Дауна (зайва 21-ша |
19 |
20 |
21 |
22 |
23 |
|
хромосома відмічена стрілкою) |
||||||
|
|
|
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
|
Рис. 5.4. Каріотип чоловіка з |
|
|
|
|
|
|
транслокаційною формою синд- |
|
|
|
|
|
|
рому Дауна (робертсонівська |
|
|
|
|
|
|
транслокація 21-ї хромосоми на |
19 |
20 |
21 |
22 |
23 |
|
14-ту; хромосома з транслока- |
||||||
цією відмічена стрілкою) |
|
|
|
|
|
|
|
|
|
|
|
|
|
79

Рис. 5.5. Синдром Дауна (сплощене обличчя, монголоїдний розріз очей, епікант, широке сплощене перенісся, макроглосія)
Рис. 5.6. Епікант у хворого з синдромом Дауна
Рис. 5.7. Чотирипальцева складка на долоні у хворого з синдромом Дауна
Таблиця 5.5. Найчастіші зовнішні ознаки
синдрому Дауна
|
Частота, % |
Вада або ознака |
від загальної |
|
кількості хворих |
|
|
Мозковий череп і обличчя: |
|
— брахіцефалія |
81,1 |
— монголоїдний розріз очей |
79,8 |
— епікант |
51,4 |
— плоска спинка носа |
65,9 |
— вузьке піднебіння |
58,8 |
— великий висолоплений язик |
? |
— деформовані вушні раковини |
43,2 |
|
|
Кістково-м’язова система, |
|
кінцівки: |
|
— низький зріст |
100,0 |
— деформація грудної клітки |
26,9 |
— короткі та широкі кисті |
64,4 |
— клинодактилія мізинців |
56,3 |
— гіпоплазія серединної |
|
фаланги 5-го пальця кисті |
|
з однією згинальною складкою |
? |
— чотирипальцева складка |
40,0 |
на долоні |
|
— сандалеподібна щілина |
? |
|
|
Очі: |
|
— плями Брушфільда |
68,4 |
— помутніння кришталика |
32,2 |
— косоокість |
? |
|
|
у 15 % — вади шлунково-кишкового тракту (атрезія або стеноз дванадцятипалої кишки, атрезія стравоходу, атрезія прямої кишки й ануса, мегаколон), у 6 % хворих — вади сечової системи. Досить часто виявляються ознаки недорозвинення зовнішніх статевих органів (крипторхізм, гіпоплазія статевого члена і мошонки), пупкові та пахові грижі, розходження прямих м’язів живота.
Розумова відсталість і затримка стато-мотор- них функцій виявляються практично в усіх хворих. Коефіцієнт розумового розвитку (IQ) у різних дітей варіює від 25 до 60. Якщо не застосовуються спеціальні методи навчання, то частіше зустрічається імбецильність (65–90 %), дебільність та ідіотія виявляються однаково часто.
Характерна затримка росту. Середній зріст дорослих хворих близько 150 см.
У хворих із синдромом Дауна спостерігаються імунодефіцитні стани, знижена репарація ДНК, через що діти з цим синдромом часто хворіють на пневмонію, важко переносять дитячі інфекції. У них значно частіше зустрічаються лейкози, ніж у здорових дітей.
Вітальний прогноз при хворобі Дауна визначається наявністю вад розвитку серцево-судинної системи і травного тракту, інфекцій дихальних шляхів (імунодефіцит вродженого характеру), хвороб крові (лейкоз) і злоякісних новоутворень, до яких схильні ці хворі. Зазвичай 20–30 % хворих гинуть на першому році життя, 50 % — в перші 5 років. За відсутності тяжких вад розвитку, при уважному догляді за хворим, своєчасній діагнос-
80
тиці та лікуванні можливих супровідних захворювань тривалість життя може досягати 50–60 років. Багато хворих з трисомією 21 здатні вести самостійне життя, опановують нескладні професії, створюють сім’ї.
Клінічна характеристика синдрому Дауна в різні вікові періоди
В період новонародженості: необхідна діагно-
стика вроджених вад серцево-судинної системи, шлунково-кишкового тракту та інших систем органів. У 3 % новонароджених є вроджені катаракти, часто зустрічається глаукома. Необхідна діагностика гіпотиреозу, який зустрічається частіше при трисомії 21 і потребує ранньої замісної терапії гормонами. Можуть бути утруднення при грудному вигодовуванні, зумовлені м’язовою гіпотонією, макроглосією. Частіше виникають запори через гіпотонічну мускулатуру кишок. Необхідно звернути увагу на можливість вродженого вивиху стегна.
Грудний вік: слід звернути увагу на схильність до судом, гіпотиреоз, інфекційні захворювання.
Дошкільний і молодший шкільний вік: серйозні вади розвитку залишаються головною причиною смерті в дитячому віці. Зберігаються схильність до інфекційних захворювань і ризик лейкозів. У хворих часто розвивається туговухість, тому рекомендується щорічна аудіометрія. Необхідні регулярні консультації окуліста, оскільки у хворих часто зустрічаються порушення рефракції, косоокість, розвиваються катаракти. У таких дітей маленькі та деформовані зуби, вони потребують регулярних оглядів стоматолога. У 15 % хворих спостерігається нестійкість атлантоаксіального з’єднання, що може призвести до стискання спинного мозку і неврологічної симптоматики. Необхідне рентгенологічне обстеження хворих перед вступом до школи. Слід звернути увагу на схильність до ожиріння.
Пубертатний вік: зберігається схильність до інфекційних захворювань і лейкозу, необхідні регулярні аудіометрії, консультації окуліста і стоматолога, контроль функції щитоподібної залози. Необхідно звернути увагу на статеве виховання підлітків. У дівчат звичайно встановлюються регулярні менструації. Більшість циклів ановуляторна, але можлива вагітність. У світовій літературі описано близько 30 випадків вагітності у жінок із синдромом Дауна. Теоретично 50 % нащадків успадковуватимуть зайву 21-шу хромосому.
Хлопці з синдромом Дауна відчувають ті ж самі статеві потяги і розлади, що і їхні однолітки. Чоловіки з синдромом Дауна, як правило, безплідні, оскільки у них порушений сперматогенез, хоча описаний один випадок зачаття дитини чоловіком з синдромом Дауна.
Шкіра дітей з синдромом Дауна схильна до сухості й екземи. Часто з’являється вугровий висип. Спостерігається гніздове облисіння.
Старший вік: хворі з синдромом Дауна старіють швидше, ніж здорові особи. У більшості хворих у старшому віці розвивається хвороба Альцгеймера. Це зумовлено тим, що один з генів хвороби Альцгеймера локалізований у 21-й хромосомі.
Медико-генетичне консультування. Для по-
Таблиця 5.6. Залежність частоти народження
дітей з синдромом Дауна від віку матері
Вік матері |
Частота народження |
||
дітей з синдромом Дауна |
|||
|
|||
|
|
|
|
До 18 років |
1 |
: 45 |
|
20 років |
1 |
: 1800 |
|
25 років |
1 |
: 1300 |
|
30 років |
1 |
: 1000 |
|
35 років |
1 |
: 300 |
|
40 років |
1 |
: 100 |
|
45 років |
1 |
: 30 |
|
49 років |
1 |
: 12 |
|
|
|
|
|
дружньої пари, що має дитину з синдромом Дауна, ризик народження ще однієї хворої дитини підвищений, він залежить від віку матері й цитогенетичного варіанта синдрому.
Встановлено тісний зв’язок між віком матері та частотою народження дітей з синдромом Дауна (табл. 5.6). Подібна залежність виявлена і при інших трисоміях за автосомами (синдроми Едвардса і Патау).
Причини цього явища не встановлені. Одна з можливих причин — особливості овогенезу в жінок. Овогенез починається в ембріональному періоді розвитку, до 7 міс проходять стадії розмноження, росту і розпочинається профаза першого поділу мейозу. В ембріональному періоді відбуваються кон’югація та кросинговер, після чого мейоз зупиняється. З підліткового віку овоцити вступають у завершальний етап мейозу. Чим більший термін від початку мейозу до його завершення, тим більше мутагенних факторів діють на організм жінок і тим більший ризик нерозходження хромосом. Проте це не пояснює високу частоту нерозходження хромосом у молодих матерів.
У разі простої трисомії та віку матері до 35 років повторний ризик народження хворої дитини не більше 1 %. Після 35 років він дорівнює подвоєному генетичному ризику для даної вікової групи.
Якщо у хворого виявлено транслокаційний варіант хвороби Дауна, то обов’язково для медикогенетичного консультування досліджують каріотипи батьків. Виявлення у когось із батьків збалансованої транслокації, що стала причиною патології у дитини, потребує при наступних вагітностях проведення інвазивної пренатальної діагностики. В цілому генетичний ризик оцінюють за спеціальними таблицями емпіричного генетичного ризику. Ризик залежить від виду транслокації та того, хто саме з батьків (мати або батько) є носієм. Ризик вищий, якщо транслокація виявлена у матері, тому що овоцити з транслокацією є життєздатнішими, ніж сперматозоїди.
Синдром Едвардса (трисомія 18)
Вперше цей синдром описав J. Edwards (1960).
Основні діагностичні ознаки: пренатальна гіпоплазія, єдина пупкова артерія, доліхоцефаль-
81

на форма черепа з навислою потилицею, мікрогенія, специфічне згинання пальців кисті, стопа-гой- далка, множинні вади внутрішніх органів, трисомія за 18-ю хромосомою.
Частота цього синдрому в популяції становить 1:5000–1:7000 новонароджених. Співвідношення хлопчиків і дівчаток дорівнює 1:3. Причини переважання серед хворих дівчаток поки що не зрозумілі.
Цитогенетичний синдром Едвардса представлений в основному простою трисомією 18, при якій у всіх клітинах виявляється додаткова хромосома. Каріотип при простій трисомній формі: 47,ХХ,+18 або 47,ХY,+18. Встановлена чітка залежність частоти народження дітей з цим синдромом від віку матері, яка навіть більш виражена, ніж при трисоміях 13 і 21. Рідко зустрічаються мозаїчні форми і як виняток — транслокаційні. Фенотипічно всі ці цитогенетичні форми не можна відрізнити.
Клінічні ознаки при синдромі Едвардса такі
(рис. 5.8):
—у новонароджених виражена гіпоплазія при нормальному терміні вагітності, середня маса при народженні 2340 г;
—маленька плацента, єдина пупкова артерія;
—характерні дизморфії обличчя і черепа: доліхоцефальна форма черепа, «нависла» потилиця, антимонголоїдний розріз очей, мікрофтальмія, низько розташовані деформовані вушні раковини, мікрогенія (маленька нижня щелепа);
—коротка груднина, вузький таз;
—характерне накладання пальців кисті — 2-й
і5-й пальці перекривають 3-й і 4-й;
—стопа-гойдалка (див. рис. 11.2), укорочений
імолоткоподібно зігнутий дорсально перший палець стопи;
—множинні вроджені вади розвитку ЦНС, сер- цево-судинної системи, органів травлення, сечової системи, статевих органів.
Прогноз завжди серйозний: 60 % хворих помирають у віці до 3 міс, однорічний вік переживає не більше 10 % хворих. У всіх тяжка розумова відсталість. Хворі гинуть внаслідок несумісних із життям вад розвитку.
Лікування неефективне, симптоматичне.
Синдром Патау (трисомія 13)
Вперше цей синдром описав K. Patau (1960).
Мінімальні діагностичні ознаки: мікроцефалія,
щілина губи і піднебіння, полідактилія, вади внутрішніх органів, трисомія 13-ї хромосоми.
Частота синдрому Патау в популяції становить 1:5000–1:7000 новонароджених. Співвідношення статей близьке до 1:1.
У 80–85 % хворих проста трисомна форма. Каріотип: 47,ХХ,+13 або 47,ХY,+13. Робертсонівські транслокації та мозаїцизм зустрічаються рідко.
Основні діагностичні ознаки при синдромі Па-
тау такі (рис. 5.9):
—черепно-лицьові дизморфії (мікроцефалія, тригоноцефалія, гіпотелоризм, щілини губи і піднебіння, дефекти скальпа, низько розташовані
йдеформовані вушні раковини та ін.);
—вади очей (мікрофтальмія або анофтальмія), носа (атрезія хоан та ін.);
—вади кінцівок (флексорне положення пальців кисті, полідактилія (рис. 5.10), «стопа-гойдалка»);
—вади внутрішніх органів: ЦНС (голопрозенцефалія, ариненцефалія), вади серцево-судинної системи, травного тракту та інших органів.
Прогноз: близько 95 % хворих з синдромом Патау гинуть протягом першого року життя, зокрема 45 % — в неонатальному періоді. Лише одиниці переживають вік більше 3 років. Усі мають тяжку розумову відсталість. Смерть настає внаслідок несумісних із життям вроджених вад розвитку.
Лікування неефективне, симптоматичне.
а |
б |
Рис. 5.8. Синдром Едвардса:
а — доліхоцефальна форма черепа, мікрофтальм, птоз, мікрогенія, низько розташовані та деформовані вушні раковини; б — характерне розташування пальців
Рис. 5.9. Синдром Патау (мікроцефалія, гіпотелоризм, серединна щілина губи і піднебіння; поєднання цих ознак дозволяє припускати голопрозенцефалію)
82
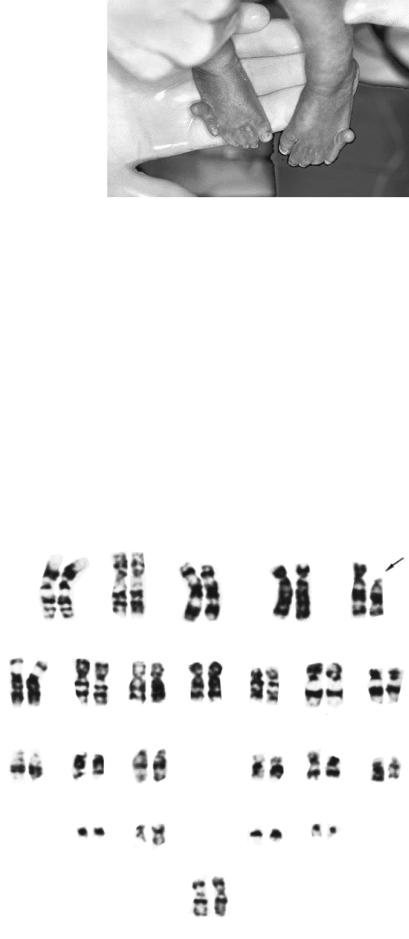
Синдром «крику кішки» (синдром «котячого крику», синдром Лежена, синдром cri du chat, синдром 5р-)
Вперше цей синдром був описаний J. Lejeune зі співавт. (1963).
Мінімальні діагностичні ознаки: незвичайний крик, що нагадує котяче нявкання, мікроцефалія, антимонголоїдний розріз очей, розумова відсталість, делеція короткого плеча 5-ї хромосоми.
Популяційна частота 1:45 000–1:50 000. Синдром обумовлений делецією короткого пле-
ча 5-ї хромосоми.
Каріотип: 46,ХХ,del 5pабо 46,XY,del 5p- (рис. 5.11).
Основні діагностичні ознаки (рис. 5.12):
—найтиповішою для цього синдрому ознакою
єспецифічний плач, що нагадує котяче нявкання або крик. Він обумовлений зміною гортані (звуження, м’якість хрящів, зменшення надгортанника, незвичайна складчастість слизової оболонки). З віком цей симптом зникає;
—м’язова гіпотонія;
—черепно-лицьові дизморфії — місяцеподібне обличчя, мікроцефалія, гіпертелоризм, широке перенісся, антимонголоїдний розріз очей, епікант, мікрогенія. Вушні раковини деформовані та низько розташовані;
—зміни дерматогліфіки;
—вроджені вади серця та інших органів;
—тяжка розумова відсталість.
Тривалість життя хворих залежить від тяжкості вроджених вад розвитку. Більшість хворих помирає в перші роки життя, близько 10 % досягають
Рис. 5.10. Двостороння полідактилія на стопах при синдромі Патау
десятирічного віку. Є одиничні описи хворих віком 50 років і старше.
Відмічається схильність до інфекційних захворювань верхніх дихальних шляхів. З віком місяцеподібне обличчя, котячий крик, м’язова гіпотонія здебільшого повністю зникають. Характерна тяжка розумова відсталість. Лікування неефективне.
Синдром Шерешевського — Тернера (45,Х)
Синдром описав у 1925 р. М. А. Шерешевський, а в 1938 р. — Г. Г. Тернер.
Мінімальні діагностичні ознаки: у новонародже-
них — лімфатичний набряк кистей і стіп, особливо помітний на нижніх кінцівках; гіпотонія, шкірні складки на шиї. У старших дітей — статевий інфантилізм, первинна аменорея, низький зріст, шкірні складки на шиї, вроджені вади серцево-судинної,
|
|
1 |
2 |
3 |
4 |
|
5 |
|
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
13 |
14 |
15 |
|
16 |
17 |
18 |
|
|
19 |
20 |
|
21 |
22 |
|
Рис. 5.11. Каріотип дівчинки |
|
|
|
|
|
|
|
з синдромом «крику кішки» |
|
|
|
|
|
|
|
(хромосома 5 з делецією корот- |
|
|
|
23 |
|
|
|
кого плеча відмічена стрілкою) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
83

Рис. 5.12. Синдром «крику кішки» (антимонголоїдний розріз очей, епікант, гіпертелоризм, місяцеподібне обличчя, низько розташовані і деформовані вушні раковини)
сечостатевої та інших систем. Повна або часткова моносомія за Х-хромосомою.
Частота у популяції синдрому 1:3000–1:3500 новонароджених дівчаток.
Це єдина форма моносомії у живонароджених. У 50 % хворих спостерігається повна форма мо-
носомії (каріотип 45,Х), у 30–40 % — мозаїчні форми (46,ХХ/45,Х), рідко ізохромосоми Х, делеції, кільцеві Х-хромосоми (рис. 5.13).
Фенотипічні прояви залежать від відсоткового вмісту аномального клону клітин.
У новонароджених і дітей грудного віку відмічаються характерні симптоми (рис. 5.14): коротка шия з надлишком шкіри, лімфатичні набряки кистей, передпліч, стіп і гомілки.
Пізніше синдром клінічно виявляється трьома групами симптомів:
1.Гіпогонадизм, недорозвинення статевих органів і вторинних статевих ознак: яєчники заміщені сполучною тканиною, фолікули відсутні, овоцити не утворюються; гіпоплазія матки і маткових труб, первинна аменорея, безплідність, недорозвинення вторинних статевих ознак (недорозвинення молочних залоз, мізерне оволосіння), недостатність естрогенів, надлишок гіпофізарних гонадотропінів.
2.Вроджені вади серцево-судинної, сечовидільної та інших систем органів зустрічаються у 25 % хворих.
3.Відставання в рості — середній зріст дорослих хворих становить 140 см.
Характерний фенотип формується вже в дитячому віці. У хворих антимонголоїдний розріз очей, епікант, низько розташовані та деформовані вушні раковини (рис. 5.15). Патогномонічним симптомом є коротка шия з крилоподібними шкірними складками (птеригіум), низький ріст волосся на шиї. Грудна клітка бочкоподібна, широко розставлені соски, множинні пігментні плями і родимки. Вторинні статеві ознаки недорозвинені. Інтелект частіше нормальний і лише у 16 % хворих — знижений.
1 |
|
2 |
3 |
|
4 |
5 |
|
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
13 |
14 |
15 |
16 |
|
17 |
18 |
|
|
|
|
|
|
|
|
Рис. 5.13. Каріотип дівчинки |
19 |
20 |
21 |
22 |
|
X |
Y |
з синдромом Шерешевського — |
|
Тернера (45,Х) |
||||||
|
|
|
|
|
|
|
84

а |
б |
Рис. 5.14. Новонароджена дівчинка з синдромом Шерешевського — Тернера:
а — шкірні складки на шиї; б — характерні лімфатичні набряки на ногах
Можуть відмічатися численні ендокринні розлади (ожиріння, цукровий діабет, тиреоїдит). Прогноз для життя сприятливий, за винятком випадків із тяжкими вродженими вадами серця і великих судин.
Лікування: у дітей — стимуляція росту, з підліткового періоду — замісна терапія жіночими статевими гормонами для формування жіночого фенотипу. Хірургічне лікування вад, косметичні операції (видалення крилоподібних складок). Описані випадки народження дітей у хворих із синдромом Шерешевського — Тернера після екстракорпорального запліднення з використанням донорської яйцеклітини.
Медико-генетичне консультування. Повторні випадки в сім’ї
синдрому Шерешевського — Тернера виключно рідкісні. Серед факторів ризику вік матері значення не має.
Синдром полісомії Х (трисомія Х, тетрасомія Х, пентасомія X)
Трисомія Х (синдром трипло-Х) вперше описана в 1959 р. у жінок з каріотипом 47,ХХХ.
Частота патології — 1:1000–1:1200 дівчаток. Серед жінок із розумовою відсталістю синдром зустрічається більш ніж в 1 % випадків.
Частіше зустрічається трисомія — каріотип 47,XXХ (повні та мозаїчні форми), рідко тетрасомія — 48,XXXХ і ще рідше пентасомія — 49,XXXXX за статевими хромосомами.
Клінічна картина трисомії Х варіабельна — від практично здорових фертильних жінок до пацієнток із вираженим гіпергонадотропним гіпогонадизмом, безплідністю, олігофренією та ін. Тяжкість захворювання корелює з кількістю Х-хромосом.
Жінки з каріотипом 47,ХХХ мають в основному нормальний фізичний і психічний розвиток (рис. 5.16). Найчастіше таких індивідів виявляють випадково при обстеженні. Це пояснюється тим, що в клітинах жінки в ембріональному періоді розвитку відбувається інактивація зайвих Х-хромосом. Як правило, не відмічається відхилень у статевому розвитку, хоча у хворих високий ризик народження дітей із хромосомною патологією або спонтанних абортів.
Рис. 5.15. Синдром Шерешевського — Тернера (антимонголоїдний розріз очей, приросла мочка вуха, шийний птеригіум, широка грудна клітка, соски молочних залоз гіпопластичні, гіпертелоризм сосків)
Рис. 5.16. Синдром трисомії Х (відсутність специфічних мікроаномалій і вад розвитку)
85

У 1/3 жінок із цим синдромом відмічається порушення репродуктивної функції (вторинна аменорея, дисменорея, рання менопауза та ін.), безплідність. Аномалії розвитку зовнішніх статевих органіввиражені незначно інеєприводомдлязвернення таких жінок до лікаря. У 2/3 хворих інтелект знижений, здебільшого незначно. У 10–15 % хворих виникають шизофренія, маніакально-депресивний психоз, епілепсія та інші психічні захворювання.
При тетрасомії Х і пентасомії Х більш виражена симптоматика, в 100 % олігофренія. Описано відхилення розумового розвитку, черепно-лицьові дизморфії, аномалії зубів, скелета і статевих органів. Проте жінки навіть з тетрасомією Х можуть бути фертильними (мають дітей).
Діагностика: у зскрібку букального епітелію виявляються два або більше тілець Барра в ядрах клітин (див. рис. 2.16, в). Остаточний діагноз встановлюють при каріотипуванні. У значної частини хворих знижений рівень естрогену і підвищений — гонадотропінів.
Лікування: симптоматичне. При гіпогонадизмі рекомендують замісну терапію жіночими статевими гормонами, при олігофренії — ноотропами.
Медико-генетичне консультування. Повторний ризик народження хворої дитини в сім’ї для сибсів менше 1 %. У фертильних хворих діти з хромосомними хворобами народжуються в 10 % випадків. Необхідна пренатальна діагностика.
Синдром Клайнфельтера
Вперше цей синдром описав Р. Клайнфельтер
(1942).
Мінімальні діагностичні ознаки: гіпогеніталізм,
гіпогонадизм у чоловіків, безплідність, каріотип
47,XXY.
Найпоширеніша хромосомна патологія у чоловіків. Частота в популяції — 1:1000 хлопчиків
(за даними Н. П. Бочкова, 2001 р. — 1:500–1:750).
Синдром обумовлений наявністю в каріотипі у чоловіка зайвих Х-хромосом. Частіше зустрічається трисомія за статевими хромосомами (рис. 5.17)
— каріотип 47,XXY (повні і мозаїчні форми), рідко тетрасомія — 48,XXXY або 48,XXYY і ще рідше пентасомія — 49,XXXXY.
У клітинах хворих з каріотипом 47,ХХY знаходять одну грудку статевого хроматину (тільце Барра), при каріотипі 48,XXXY — дві грудки і при каріотипі 49,XXXXY — три.
Тяжкість захворювання корелює з кількістю додаткових Х-хромосом.
При трисоміях перебіг хвороби може бути легким. До періоду статевого дозрівання хлопчики зазвичай розвиваються нормально, інтелект без відхилень або з незначним відставанням у психічному розвитку. Синдром частіше виявляється клінічно в період статевого дозрівання у вигляді недорозвинення сім’яників і вторинних чоловічих статевих ознак.
Хворі з синдромом Клайнфельтера високі на зріст (рис. 5.18), з диспропорційно довгими кінцівками, євнухоїдною статурою, оволосінням за жіночим типом (брак рослинності на обличчі, горизонтальний рівень росту волосся на лобку). У 30 % хворих спостерігається гінекомастія (розвиток молочних залоз). Існує ризик злоякісної пухлини
1 |
2 |
|
3 |
|
4 |
5 |
|
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
13 |
14 |
15 |
|
16 |
17 |
18 |
|
|
|
21 |
22 |
|
X |
Y |
Рис. 5.17. Каріотип чоловіка |
19 |
20 |
|
з синдромом Клайнфельтера |
||||
|
|
|
|
|
|
|
(ХХY) |
86

Рис. 5.18. Синдром Клайнфельтера (евнухоїдна статура, непропорційно довгі кінцівки, горизонтальний ріст волосся на лобку, нормальні розміри статевого члена, мікроорхідизм)
молочних залоз. Зовнішні статеві органи розвинені за чоловічим типом, крипторхізм зустрічається рідко. Внутрішні статеві органи гіпоплазовані. Характерним є мікроорхідизм (малі розміри яєчок) і нормальні розміри статевого члена. Гістологічно виявляють дегенерацію гермінативного епітелію та гіаліноз сім’яних канатиків. Хворі, як правило, безплідні (азооспермія, олігоспермія). Інтелектуальний розвиток хворих не змінений або наявна олігофренія.
Лікування: замісна терапія андрогенами з 10– 12 років. Для стимуляції росту волосся на обличчі використовують креми та мазі з андрогенами. Гінекомастія іноді потребує хірургічної корекції.
Медико-генетичне консультування. Ризик для сибсів менше 1 %. Ризик народження хворих дітей у пацієнтів з синдромом Клайнфельтера у разі збереження фертильності становить 10 %. Необхідна пренатальна діагностика.
Синдром полісомії Y-хромосоми
Цей синдром вперше описали Сандберг і співавт. (1961).
Частота в популяції — 1:1000 новонароджених хлопчиків і 1:10 серед чоловіків із зростом більше
2 м.
Каріотип хворих 47,ХYY, рідше 48,ХYYY; 49,ХYYYY або мозаїцизм (45,Х/49,ХYYYY та ін.).
Клінічні симптоми варіюють від практично нормальних чоловіків за фізичним і розумовим розвитком до пацієнтів з легкою розумовою відсталістю, схильністю до агресивних і навіть кримінальних вчинків.
Надмірний ріст починається в дитячому віці, у дорослих він становить у середньому 186 см. Кожна Y-хромосома збільшує зріст приблизно на 15 см. Іноді виявляють акромегалоїдні риси — збільшення нижньої щелепи, кистей, стіп, грубі риси обличчя, виступаючі надбрівні дуги (рис. 5.19).
Статева функція у багатьох хворих нормальна, фертильність збережена (іноді гіперсексуальність).
Рис. 5.19. Синдром полісомії Y (високий зріст, акромегалоїдні риси — збільшення нижньої щелепи, кистей, стіп, грубі риси обличчя, виступають надбрівні дуги, інтелект нормальний, у деяких випадках може бути легка розумова відсталість, схильність до агресивних і навіть кримінальних вчинків)
87
У тяжких випадках наявні крипторхізм, порушен- |
1. Вони мають чітку клінічну картину, оскіль- |
ня сперматогенезу, безплідність. |
ки мікроделеція або мікродуплікація порушують |
У 30–40 % хворих легка розумова відсталість, |
маленьку ділянку хромосоми (часто один ген). |
зниження критики, агресивність, вибуховість, збо- |
2. Інколи ці синдроми можуть бути зумовлені |
чення потягів. Навіть при нормальному інтелекті |
не тільки хромосомною аберацією, але і генними |
часті істероподібні прояви в поєднанні з вибуховіс- |
мутаціями зазначеного гена. Наприклад, ретино- |
тю, конфліктністю, недостатньою критикою. |
бластома може бути зумовлена делецією ділянки |
Мікроознаки: макроцефалія, високе перенісся, |
довгого плеча 13-ї хромосоми (q14) або точковою |
«готичне» піднебіння, порушення росту зубів, мак- |
мутацією зазначеного гена. |
роотія, лійкоподібна груднина, вальгусна дефор- |
3. Причиною розвитку деяких синдромів може |
мація ліктьових і колінних суглобів, перших паль- |
бути не тільки мікроделеція, але й однобатьківсь- |
ців стіп, радіоульнарний синостоз. |
ка дисомія та порушення геномного імпринтингу |
Лікування: симптоматичне. |
(синдроми Ангельмана і Прадера — Віллі). |
Медико-генетичне консультування. Ризик для |
4. Зустрічаються рідко (у більшості випадків |
сибсів менше 1 %. Ризик народження хворих дітей |
1:50 000–1:100 000 у новонароджених). |
у пацієнтів з цим синдромом у разі збереження |
5. Для діагностики використовують молекуляр- |
фертильності — 10 %. Необхідна пренатальна діаг- |
но-цитогенетичні методи (FISH-метод). Звичайне |
ностика. |
каріотипування виявляється неефективним. |
|
|
Синдром Ангельмана |
|
|
|
(синдром «щасливої ляльки») |
|
5.7. ПОНЯТТЯ ПРО |
Цей синдром вперше описав H. Angelman |
||
МІКРОЦИТОГЕНЕТИЧНІ |
(1965). |
||
Мінімальні діагностичні ознаки: тяжка розумо- |
|||
СИНДРОМИ |
|
||
|
ва відсталість, груба затримка мовного розвитку, |
||
|
|
судоми, характерна хода, немотивований сміх. |
|
До цієї групи хромосомних хвороб входять |
Частота в популяції 1:50 000. |
||
синдроми, зумовлені делеціями або дуплікаціями |
Синдром обумовлений мікроделецією ділянки |
||
дуже маленьких ділянок хромосом. Їх відповідно |
довгого плеча 15-ї хромосоми (q11-q13) материнсь- |
||
називають мікроделеційними і мікродуплікаційни- |
кого походження (рис. 5.20). Він може бути також |
||
ми синдромами. Основні відомості про мікроци- |
наслідком уніпарентної дисомії (успадкування |
||
тогенетичні синдроми наведено в табл. 5.7. |
двох хромосом 15 від батька, тобто материнські |
||
Для мікроцитогенетичних синдромів характер- |
гени відсутні). У розвитку клініки має значення ге- |
||
не таке: |
|
номний імпринтинг. |
|
|
Таблиця 5.7. Мікроцитогенетичні синдроми |
||
|
|
|
|
Назва синдрому |
Залучена ділянка хромосо- |
Основні симптоми |
|
або хвороби |
ми (делеція або дуплікація) |
||
|
|||
|
|
|
|
|
Мікроделеційні синдроми |
||
|
|
|
|
Ретинобластома |
13q14.1-q14.2 |
Пухлина сітківки (одноабо двостороння) |
|
|
|
в дитячому віці |
|
|
|
|
|
Синдром Ді Джорджі |
22q11.21 |
Судоми (гіпокальціємічні), аплазія або гіпоплазія |
|
|
|
тимуса, дизморфії лицьового черепа, вади серця |
|
|
|
|
|
Синдром Ангельмана |
15q11-q13 |
Незвичайне обличчя, атаксія, гіпотонія, епілепсія, |
|
|
у хромосомі від матері |
пароксизми сміху, мікроцефалія, відсутність мови |
|
|
|
|
|
Синдром Прадера — |
15q11-q13 |
Ожиріння тулуба і проксимальних відділів кінцівок, |
|
Віллі |
у хромосомі від батька |
дизморфії лицьового черепа, гіпотонія, |
|
|
|
гіпогонадизм, розумова відсталість, маленькі |
|
|
|
кисті і стопи |
|
|
|
|
|
Пухлина Вільмса |
11р13 |
Нефробластома |
|
|
|
|
|
|
Мікродуплікаційні синдроми |
||
|
|
|
|
Синдром Беквіта — |
|
Грижа пупкового канатика, макроглосія, гігантизм, |
|
Відеманна |
11р15 |
гіпоглікемія, мікроцефалія, вроджені вади |
|
|
|
внутрішніх органів |
|
|
|
|
|
88