

Влияние изменения реакционной способности функциональных групп на зависимости степени превращения мономера и степени полимеризации от глубины протекания процесса. Реакционная способность мономеров.
Реакционная способность реагентов при поликонденсации во многих случаях определяет как ход процесса, так л молекулярную массу полимера. Поэтому очень важно определить реакционную способность реагентов поликонденсационного процесса, в том числе мономера.
Особенности взаимодействия бифункциональных мономеров.
Реакция бифункционального мономера с монофункциональным протекает в две стадии: сначала реагирует одна функциональная группа, затем вторая. Например:
29
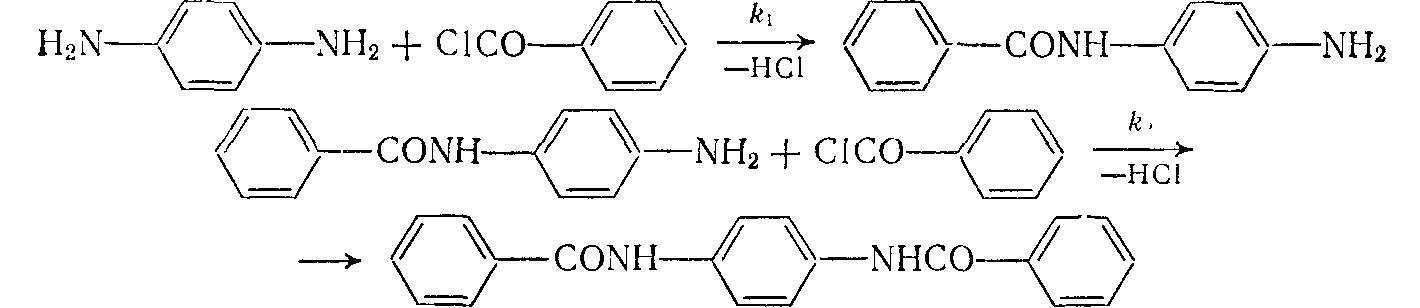
Для многих мономеров алифатического ряда константы скорости реакций k1иk2равны между собой и изменение числа моно- и дизамещенных мономеров в ходе процесса определяется только концентрационными факторами.
Для мономеров ароматического ряда и некоторых мономеров алифатического ряда k1k2. В этом случае кроме концентрационных факторов большую роль играет отношениеk1/k2. Величина, характеризующая относительную реакционную способность второй функциональной группы по сравнению с первой, называется коэффициентом взаимозависимости функциональных групп. Он показывает, во сколько раз изменилась реакционная способность второй функциональной группы после вступления в реакцию первой. От величиныk1/k2 зависит соотношение моно- и дизамещенных продуктов. Отношениеk1/k2> 1 благоприятствует образованию монозамещенных; отношениеk1/k2<. 1 благоприятствует образованию дизамещенных (при неполной конверсии функциональных групп).
Закономерности по стадийного взаимодействия бифункциональных мономеров ароматического ряда были подробно изучены на примере ароматических диаминов и дихлорангидридов ароматических дикарбоновых кислот.
Было установлено, что в изученных соединениях отношение k1/k2зависит какorприроды самого бифункционального мономера, так и от природы присоединяемого к нему фрагмента. Так, при ацилировании З-хлорбензоилхлоридом бензидинаk1/k2. = 4.8, а 4,4-диаминодифенилметанаk1/k2 = 1.3; при взаимодействии дихлорангидрида терефталевой кислоты с п-хлоранилиномk1/k2 =1.35, а при взаимодействии с п-толуидиномk1/k2 = 2.45. Зависимостьlg(k1/k2) от индукционной константы заместителя является линейной.
30
Было показано также, что отношение k1/k2не зависит от применяемого растворителя. При взаимодействии бифункциональных мономеров несимметричного строения, например
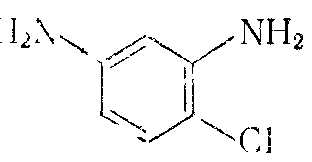
реакционная способность аминогрупп будет различаться с самого начала; кроме того, она изменяется еще и в ходе процесса.
Возможен случай, когда k1/k2< 1, т. е. когда активность вторичной группы выше активности первой. Так, по некоторым данным отношениеk1/k2. для реакции дихлорангидрида терефталевой кислоты с п-нитрофенолом (катализатор—триэтиламин) равно 0.77. Подобные пары мономеров весьма интересны, поскольку их поликонденсация имеет ряд особенностей (см. рис. 2.3 и ниже).
Все изложенное выше относилось к реакциям бифункциональных соединении с монофункциональными. При взаимодействии двух бифункциональные соединений начало процесса характеризуется четырьмя константами скорости. Причем различие между k1иk4может быть весьма значительным. Так, для ароматических диаминов и дихлорангидридов ароматических дикарбоновых кислот значениеk1/k4может изменяться от 6 до 73. Посколькуk4характеризует реакцию между димерами,k2иk3—между мономерами и димерами иk1—между мономерами, то соотношение между этими константами определяет характер процесса—относительную скорость взаимодействия в нем мономеров и олигомеров. Это следует учитывать при рассмотрении закономерностей поликонденсационных процессов.(см. рис. 2.3, кривые 3 и 4).
При различии в активностях функциональных мономеров и олигомеров (например, при разной активности первичной и вторичной групп мономеров) вид зависимости n=f(x) может измениться. Еслиk1/k2>l, то зависимостьn=f(x) близка к зависимости, описываемой уравнением (2.1). Еслиk1/k2<l, то эта зависимость сильно отличается от «классической», и тем больше, чем меньше отношениеk1/k2. При очень малых отношенияхk1/k2становится возможным получение высокомолекулярных продуктов при гораздо меньших степенях превращения.
31
На рис. 2.7 представлены зависимости степени поликонденсации (вернее, lgn) от глубины процесса для различных соотношений активностей первичных и вторичных групп мономеров. Из рис. 2.7 видно, что приk1/ki= 10-3(кривая 1) полимер со степенью поликонденсации 100 (lg = 2) получается при глубине процесса х = 0.75 вместо 0.99 согласно уравнению (2.1) (кривая 5) иk1/ki= 1 (кривая 4).
Изменение активности реагирующих компонентов системы в ходе процесса может происходить, например, при поликонденсации ароматических мономеров с взаимозависимыми функциональными группами, когда активность второй группы меняется после вступления первой в полимерную цепь. Интересным примером изменения активности при k1/k2<lявляется каталитическое взаимодействие дихлорангидрида терефталевой кислоты с п-нитрофенолом. Кстати, избирательный катализ реакций одних реагентов (например, сn= 2) также может явиться причиной изменения активностей между реагентами системы со всеми вытекающими из этого последствиями.
РИС. 2 7. Зависимость
логарифма степени поликонденсации от
глубины процесса при различных константах
скорости первичных и вторичных групп
мономеров (расчет на ЭВМ): 1 – k1/ki= 10-3, 2 –k1/ki= 10-2, 3 –k1/ki= 10-1, 4 –k1/ki= 1, 5 – кривая рассчитана по уравнению
(2.1).

Изменение активности функциональных групп можно учесть введением дополнительного параметра в уравнение (2.1). Тогда уравнение (2.1) примет вид
n= 1/(1 —х) (2.3)
32
где —коэффициент, учитывающий изменение активности функциональных групп по ходу процесса: увеличению активности олигомеров соответствует (по сравнению с мономером)> 1, уменьшению< 1.
Для определения уравнение (2.3) следует переписать в форме
1/n= 1-x+C, где С — константа (2.4)
Это уравнение переходит в уравнение (2.1) при = 1 и С == 0. Как видно из рис. 2.9, уравнение (2.4) более точно описывает опытные данные, чем уравнение (2.1): экспериментальные точки хорошо ложатся на график зависимости, описываемой уравнением (2.4) при х == 0.68 и С = -0,33. Следует учитывать, что уравнение 2.4)может описать только часть процесса, но не весь процесс полностью.
Уравнение (2.4) может описывать и другие сложные процессы поликонденсации, причем и С в этом случае могут играть роль эмпирических коэффициентов, позволяющих получить кривую, наиболее точно описывающую экспериментальные данные (в настоящее время различные варианты поликонденсационных процессов следует моделировать и рассчитывать на ЭВМ.
Следует, конечно помнить, что все приведенные выше уравнения (2.1) — (2.4) применимы для поликонденсации, не осложненной диффузионными факторами. При диффузионном торможении процесса (например, при поликонденсации в гетерогенных системах) закономерности, связывающие степень поликонденсации с глубиной превращения, будут иными (см. лекционный материал).