Matta, Boyd. The quantum theory of atoms in molecules
.pdf31211 Topological Analysis of Proteins as Derived from Medium and High-resolution Electron Density
11.5.3
Electrostatic Interaction Energies
Electrostatic interaction energy values were calculated between the adenine group of NADPþ and the adenine binding site. All three models were considered, at atomic resolution and using a smoothing degree of t ¼ 0:420 A˚ 2. The atomic charges of the adenine group were those of MacKerell et al. [55]. Interestingly, interaction energy values obtained within the framework of a simple point charge Coulombic interaction depict, in all models, stabilization of the adenine group within the protein cavity. Indeed, electrostatic interaction energies computed in
the atomic description using QTAIMEXP charges and multipolar database QVAL charges are 152.3 and 84.3 kJ mol 1, respectively, whereas the fragment description leads to 74.6, 49.8, and 46.5 kJ mol 1 with QTAIMEXP, QVAL and unit charges, respectively. This stabilization is emphasized in the framework of the QTAIMEXP charge model for both atomic and medium-resolution descriptions, because of to their greater magnitudes. Surprisingly, interaction energy obtained for the unit-charge model is very close to that obtained for QVAL fragment charges, despite discrepancies observed in electrostatic potential representations.
11.6
Conclusions and Perspectives
This chapter presents several concepts and techniques for conducting topological analysis of protein ED distributions at di erent levels of resolution. It particularly focuses on the transferability of electronic information from high to lowresolution representations.
Several descriptions of ED distributions were considered. First, experimental ED distributions were modeled at a subatomic resolution level (d < 0:9 A˚ ) by use of a multipolar description that enables determination of atomic charges in accordance with the Bader QTAIM approach [24, 36]. Second, a CP analysis technique developed by Johnson [26] was applied to both experimental and promolecular ED distributions at medium resolution (d A3 A˚ ) to locate their ED maxima. In this second approach, promolecular ED grids were obtained by Fourier transformation of calculated structure factors. Third, a smoothing algorithm was applied to a Gaussian promolecular description of the ED distribution to locate its maxima and to define their corresponding molecular fragments by a clustering procedure.
Application of these di erent approaches were proposed to study the electrostatic properties of the adenine binding site of the hAR structure bound with the cofactor NADPþ [6]. More precisely, the atomic charges that were obtained at a subatomic resolution level were used to calculate medium-resolution fragment charges. Two sets of atomic charges, characterized by overall charge neutrality, were used – database atomic charges directly obtained from the valence popula-

11.6 Conclusions and Perspectives 313
tions and topological QTAIM charges derived from the transferred ED multipolar database.
Topological analysis of ED distributions reconstructed from the multipolar database showed that atomic properties such as charge and volume enabled classification of the atoms according to their chemical function or their chemical environment. For example, atoms bonded to O and N, or to C, formed di erent clusters. This observation emphasized the e ect of the electronegativity of atoms involved in a chemical bond. This clustering of topological properties was described in more detail for the atoms belonging to the peptide group HNaHaCa aCbO, and to those forming the aromatic group of the amino acid tyrosine. The QTAIM charges seemed to be totally transferable and were thus regarded as simple point charges in electrostatic property calculations.
Molecular electrostatic potentials generated by the adenine binding site in the hAR structure and the electrostatic interaction energies of the adenine moiety with the protein binding site were calculated using several charge models. The two first models were built from the two sets of atomic charges. Each of these two sets was also used to calculate fragment charges. Finally, a fifth charge model was built by assigning formal unit charges to the ARG, LYS (þ1) and GLU, ASP ( 1) side-chains. Comparison of these five models led to several observations. First, although atomic database and QTAIM charges have similar signs, the QTAIM charges have more pronounced positive and negative values than the database charges. When applied to fragment representations, database and QTAIM fragment charge signs may di er, depending on fragment content. Second, all charge models, atomic or fragment-based, gave similar results, i.e. the adenine moiety is located in a slightly positive area and its aNH2 and H8A group/atom are pointing toward a negative area. It is however shifted away from the adenine group with the topological model when t increases. Fragment representations, up to medium smoothing (t ¼ 0:420 A˚ 2), are thus acceptable descriptions for modeling electrostatic properties of a protein site. Third, two positive regions occur on either side of the adenine plane, except with the formal unit charge model. In that last instance all four unit charges of the adenine binding site are located on one side only of the adenine rings and do not properly depict its 3D electrostatic properties. Finally, all electrostatic interaction energies adopt the same stabilization e ect toward the adenine group within its binding site, even with the formal unit charges. The topologically based electrostatic energy is, however, more negative, because of the more pronounced individual atomic charges.
This work will have a variety of applications. In this chapter, the clustering procedure used to partition a protein structure into fragments is based on progressive smoothing of the protein promolecular ED distribution, i.e. a change in the overall temperature factor. The decomposition of a protein structure as a function of the crystallographic resolution rather than the temperature factor could be considered. This would enable deeper comparison with the CPs obtained using Johnson’s approach [26]. For more general application purposes, a database of atomic charges could be combined with a database of protein fragment contents and used to automatically generate reduced protein steric and electrostatic representa-

|
|
|
References |
315 |
|
|
|
|
|
20 |
Pearlman D. A.; Case D. A.; Caldwell |
38 |
Pichon-Pesme V.; Lachekar H.; |
|
|
J. W.; Ross W. S. et al.; Comp. Phys. |
|
Souhassou M. and Lecomte C. Acta |
|
|
Commun. 91 (1995), 1–41. |
|
Cryst. B 56 (2000), 728–737. |
|
21 |
Koritsanszky T.; Volkov A. and |
39 |
Matta C. F. and Bader R. F. Proteins |
|
|
Coppens P. Acta Cryst. A 58 (2002), |
|
Struct. Funct. Genet. 52 (2003), 360– |
|
|
464–472. |
|
399. |
|
22 |
Dittrich B.; Koritsanszky T. and Lu¨ger |
40 |
Frisch M. J.; Trucks G. W.; Schlegel |
|
|
P. Angew. Chem. Int. Ed. 43 (2004), |
|
H. B.; Gill P. M. W. et al.; (1995) |
|
|
2718–2721, and references cited |
|
Gaussian94, Revision B.3. Gaussian |
|
|
therein. |
|
Inc., Pittsburgh PA, USA. |
|
23 |
Bader R. F. W. (1990) Atoms in |
41 |
Wiest R.; Pichon-Pesme V.; Be´nard |
|
|
Molecules: A Quantum Theory. Oxford |
|
M. and Lecomte C. J. Phys. Chem. 98 |
|
|
University Press, Oxford, UK. |
|
(1994), 1351–1362. |
|
24 |
Souhassou M. and Blessing R. H. |
42 |
Souhassou M.; Lecomte C.; Blessing |
|
|
J. Appl. Cryst. 32 (1999), 210–217. |
|
R. H.; Aubry A. et al.; Acta Cryst. B 47 |
|
25 |
Hall S.; du Boulay D. and Olthof- |
|
(1991), 253–266. |
|
|
Hazekamp R. (2002) The Gnu XTAL |
43 |
Souhassou M.; Lecomte C.; |
|
|
System of Crystallographic Software, v. |
|
Ghermani N.-E.; Rohmer M.-M. et |
|
|
3.7.2, http://xtal.sourceforge.net/. |
|
al.; J. Am. Chem. Soc. 114 (1992), |
|
26 |
Johnson C. K. (1977) ORCRIT: The |
|
2371–2382. |
|
|
Oak Ridge Critical Point Network |
44 |
Pichon-Pesme V. and Lecomte C. Acta |
|
|
Program. Oak Ridge TN, USA. |
|
Cryst. B 54 (1998), 485–493. |
|
27 |
Leherte L. and Vercauteren D. P. |
45 |
Dahaoui S.; Jelsch C.; Howard J. A. K. |
|
|
J. Mol. Model. 3 (1997), 156–171. |
|
and Lecomte C. Acta Cryst. B 55 |
|
28 |
Girone´s X.; Amat L. and Carbo´-Dorca |
|
(1999), 226–230. |
|
|
R. J. Chem. Inf. Comput. Sci. 42 |
46 |
Benabicha F.; Pichon-Pesme V.; |
|
|
(2002), 847–852. |
|
Jelsch C.; Lecomte C. et al.; Acta |
|
29 |
Amat L. and Carbo´-Dorca R. J. |
|
Cryst. B 56 (2000), 155–165. |
|
|
Comput. Chem. 18 (1997), 2023–2039. |
47 |
Guillot R.; Muzet N.; Dahaoui S.; |
|
30 |
Kostrowicki J.; Piela L.; Cherayil B. J. |
|
Lecomte C. et al.; Acta Cryst. B 57 |
|
|
and Scheraga H. A. J. Phys. Chem. 95 |
|
(2001), 567–578. |
|
|
(1991), 4113–4119. |
48 |
Bouhmaida N. (1993) Ph. D. Thesis, |
|
31 |
Leherte L. Acta Cryst. D 60 (2004), |
|
LCM3B Universite´ Henri Poincare´, |
|
|
1254–1265. |
|
France. |
|
32 |
Leherte L.; Dury L. and Vercauteren |
49 |
Pichon-Pesme V. (2001) Personal |
|
|
D. P. J. Phys. Chem. A 107 (2003), |
|
communication. |
|
|
9875–9886. |
50 |
Minichino A.; Habash J.; Raftery J. |
|
33 |
Leung Y.; Zhang J.-S. and Xu Z.-B. |
|
and Heliwell J. R. Acta Cryst. D 59 |
|
|
IEEE T. Pattern Anal. 22 (2000) |
|
(2003), 843–849. |
|
|
1396–1410. |
51 |
Becue A. (2004) Ph. D. Thesis, |
|
34 |
Guillot B.; Jelsch C.; Podjarny A.; |
|
Faculte´s Universitaires Notre-Dame |
|
|
Lecomte C. Acta Cryst. D. To be |
|
de la Paix (FUNDP) Namur, Belgium. |
|
|
published. |
52 |
Guillot B.; Muzet N.; Artacho E.; |
|
35 |
Yabe-Nishimura C. Pharmacol. Rev. 50 |
|
Lecomte C. et al.; J. Phys. Chem. B |
|
|
(1998), 21–34. |
|
107 (2003), 9109–9121. |
|
36 |
Souhassou M. (1997) NEWPROP: |
53 |
Cunane L. M. and Taylor M. R. Acta |
|
|
Computer program to calculate the |
|
Cryst. D 53 (1997), 765–776. |
|
|
topological properties of electron density. |
54 |
Ghermani N.; Lecomte C. and |
|
|
Internal Report, LCM3B, Universite´ |
|
Bouhmaida N. Z. Naturforsch. A 48 |
|
|
Henri Poincare´, France, http:// |
|
(1993), 91–98. |
|
|
www.lcm3b.uhp-nancy.fr. |
55 |
MacKerell Jr. A. D.; Wiorkiewicz- |
|
37 |
Pichon-Pesme V.; Lecomte C.; Wiest |
|
Kuczera J. Jr. and Karplus M. |
|
|
R. and Be´nard M. J. Am. Chem. Soc. |
|
J. Am. Chem. Soc. 117 (1995), 11946– |
|
|
114 (1992), 2713–2715. |
|
11975. |
|

317
12
Fragment Transferability Studied Theoretically and Experimentally with QTAIM – Implications for Electron Density and Invariom Modeling
Peter Luger and Birger Dittrich
12.1 Introduction
It is interesting to note that as early as 1915, three years after the discovery of X- ray di raction, Peter Debye made a note that the distribution of electron density should be obtainable from this new experimental method [1]. It took ninety years, however, until, in the autumn of 2005, Philip Coppens’ highlight article (entitled ‘‘Charge Densities Come of Age’’) appeared in ‘‘Angewandte Chemie’’ [2]. He pointed out it was a long way from Debye’s vision in 1915 to the current stage of electron-density work, that progress was slow, and that the major advances had all occurred in the last decade. The substantial progress made in electron density research in the last few years rests on three pillars:
1.advances in experimental techniques – high-brilliance X-ray sources (synchrotron beamlines), very low temperatures
(T ! 10 K), and CCD area detection;
2.theoretical developments, e.g. Bader’s QTAIM theory [3], enabling the derivation of quantitative topological data; and
3.computing – the development and distribution of specific computer program systems for all aspects of electronic density work, including refinement, analysis, and visualization of results.
As a result of these simultaneous developments, electron-density studies of entire classes of chemically related compounds or of larger molecules became feasible in a reasonable time. Routine application of electron-density work is in sight.
In this chapter we would like to start with a brief summary of recent experimental advances which enable verification of the transferability of atomic fragments from significant experimental data, a key feature of Bader’s QTAIM theory. Understanding of what constitutes chemical similarity led to the introduction of invarioms, pseudoatom fragments of electron density that are invariant in transfer from one molecule to another, and to their recent applications. Invarioms en-

318 12 Fragment Transferability Studied Theoretically and Experimentally with QTAIM
able replacement of the independent atom model (IAM), and their introduction will possibly add another pillar to high-resolution crystallographic work.
12.2
Experimental Electron-density Studies
12.2.1
Experimental Requirements
The electron density of a chemical structure consists of a large spherical contribution and a very small nonspherical contribution located mainly in the regions of the covalent bonds and some nonbonding (lone pair) regions. Because chemistry happens in the latter regions, one is more interested in the aspherical part of the electron density and observation of these small e ects requires very precise experiments. Atoms in crystals are not at rest, being described by well-known displacement terms which decrease as the temperature is reduced. Because thermal smearing should be as small as possible, data collection should be performed at the lowest temperature achievable. At low temperatures, moreover, high-order reflections are more likely to have significant intensities above the background. High-order data are needed not only to improve accuracy and resolution but also to provide su cient data for the refinement of the increased number of variables of the Hansen and Coppens multipole model [4]. To summarize, several experimental requirements must be met:
|
excellent single-crystal quality |
|
precise intensity-data collection |
|
high resolution (sin y=l > 1:0 A˚ 1 or d < 0:5 A˚ ) |
|
hard X-radiation, e.g. MoKa or shorter, synchrotron radiation |
|
high completeness and redundancy in reciprocal space, |
|
hence use of an area detector is preferable |
|
low temperature (T a100 K) |
Whether or not a data set is suitable for electron-density determination must be carefully examined. Several criteria can be used:
conventional figures of merit, Rint, Rs, R(F) or R(F2), Rw(F), GoF
the Hirshfeld test [5] – a covalently bonded pair of atoms of comparable mass can be regarded as rigid with respect to thermal motion, so that the displacement parameter components of the atoms in question should be equal in bond direction
the quality of the residual electron density obtained by Fourier transformation of the di erence between observed and multipole-model structure factors – the residual density should be unstructured and low
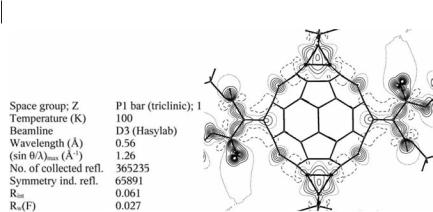
320 12 Fragment Transferability Studied Theoretically and Experimentally with QTAIM
Fig. 12.2 High-resolution synchrotron data set for the fullerene derivative dodecakisethoxycarbonyl-C60-fullerene. Left: summary of the crystallographic data. Right: static deformation density map in an equatorial plane of the C60 sphere [9] Copyright 2002 and Reproduction with Permission from American Chemistry Society.
properties. Most important for electron density experiments are the very high intensity and the tunable wavelength, enabling choice of l A0:5 A˚ or even shorter. This enables high-resolution data sets to be collected, if needed also for smaller crystals, for which absorption and extinction problems are minimized.
An example in which synchrotron radiation was essential for collection of a high-resolution data set is illustrated by Fig. 12.2 for a highly substituted C60 fullerene derivative.
Fullerenes are usually very unsuitable for experimental electron-density studies because of poor crystal quality and the high mobility of the molecules in the crystal lattice. This is why, for example, there is no electron-density study of an unsubstituted fullerene. After several unsuccessful attempts we were able to grow suitable crystals of the highly substituted Th-symmetrical derivative dodecakise- thoxycarbonyl-C60-fullerene, C102H60O24 [10] (Fig. 12.2), which co-crystallized with 1,2-difluorobenzene. On the basis of a data set of more than 350,000 reflections measured in five days at 100 K up to a resolution of sin y=l ¼ 1:26 A˚ 1 on beamline D3 of Hasylab/DESY, a properly resolved electron-density distribution and related bond-topological and atomic properties [9, 11] were derived.
If well-di racting crystals of su cient size can be grown, high-resolution data collection can be conducted with laboratory equipment. An example is a data set for vitamin B12 (Fig. 12.3). Although B12 crystals of desired size can be grown rather easily in a variety of solvents, they are extremely unstable when taken out of the mother liquor, being destroyed within seconds. This made careful, but also time-consuming crystal preparation necessary. To avoid the risk of wasting synchrotron beamtime, data were collected by use of a Bruker Smart 1K di ractometer with conventional MoKa radiation from a sealed 2.4-kW tube and were very suitable for further electron-density evaluation. Figure 12.3 shows the static defor-