

Matta, Boyd. The quantum theory of atoms in molecules
.pdf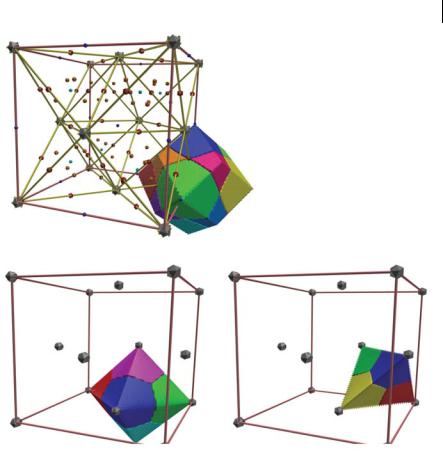
8.3 Crystalline Isostructural Families and Topological Polymorphism 213
Fig. 8.2 Six Al–c1 and eight Al–c2 primary bundles form the atomic basin of Al. Four and six bundles, respectively, form the repulsion basin of c1 and c2 cage points. Each bundle is of a single solid color to show their matching in forming the coarser basin objects.
8.3
Crystalline Isostructural Families and Topological Polymorphism
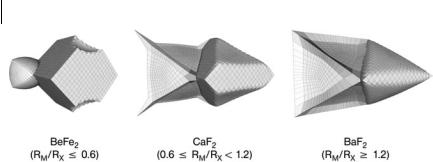
A most significant fact emerges when the electron density is systematically analyzed in families of crystals – compounds with the same crystalline structure which are expected to have similar chemical properties have, however, di erent topologies of r. An example of this topological polytypism is provided by the alka- line-earth halides, MX2, in the fluorite structure. These 20 compounds have one of three di erent topologies, represented in Fig. 8.3. What is most interesting is that the actual topology of a compound can be exactly predicted by determining the size ratio of the M and X species along the M–X bond path. When the M cation is less that 0.6 times the X anion, large faces created by the X–X bond critical
214 8 Topology and Properties of the Electron Density in Solids
Fig. 8.3 Topological polytypism in alkaline-earth fluorites.
points dominate the anionic basin, which fills most of the crystal volume. The X–X bond paths continue to exist when RM is in between 0.6 and 1.2 times RX , but the size and e ect of the X–X bonds decays progressively as we approach the upper limit. When RM=RX b1:2, both M and X basins finally approach the shape dictated by their coordination in the crystalline structure (an octahedron and a tetrahedron, respectively) and the only bonding remaining is the M–X bond path between the nearest neighbors.
Similar geometrical reasoning explains the topological polytypism in other families we have examined, for example the B1 and B2 phases of alkali halides [33, 34], the alkaline/alkaline-earth perovskites AMX3 [35], and their elpasolites A2MX4 [36]. The distance from a nucleus to its basin surface is very dependent on direction, but those directions determined by the bond paths contain the most useful information and enable us not only to systematize the occurrence of a given topology but also to explain trends in the thermodynamic properties of the crystals [37].
The examples above are of di erent compounds, albeit structurally related, with di erent topologies. A change of crystal structure as a result of a phase transition will also modify the electron density topology, but the interesting question is whether is it is possible for a single compound to have several di erent topologies without there being a structural change. We have found two di erent types of situation in which this behavior seems to be the norm.
The first example is the metallic phases of the low-electronegativity elements. BCC Li is the most extreme example, because it has approximately twenty di erent topologies for a change in the lattice size from 2.4 to 4.8 A˚ [18], but all the alkaline and alkaline-earth metals share the same tendency toward topological change. Perhaps most amazing is that the change follows a well defined trend, revealed when the many topologies are classified according to some significant features:
B2: first and second metal neighbors are bonded; B1: only nearest neighbors bond CPs remain; and
M: the topology has nonnuclear maxima (NNM), also referred to as nonnuclear attractors (NNA), in addition to the prototypical nuclear ð3; 3Þ CPs [38–40].

8.4 Topological Classification of Crystals 215
The topology of the di erent metals follows a common trend on compression: B2 ! B1½! M& ! B2 . . . . The cause of this topological lability is that the electron density is almost constant, i.e. flat, throughout the valence region of these metals. An appropriate measurement of the valence flatness is provided by the ratio [41]:
rmin
f ¼ c ð3Þ
rbmax
where rcmin is the absolute minimum of the electron density, and rbmax is the maximum electron density found among the bond CPs. Typical covalent, ionic, and molecular solids have f values quite close to zero. Alkali metals, in contrast, approach f ¼ 100% (98% Li, 95% Na and K, 91% Rb, and 88% Cs at their respective experimental geometries) and truly resemble the ideal Drude model of a free electron gas.
Topological polymorphism can also be expected for nonelementary compounds when the di erent elements share an extremely similar electronegativity. BP is a prototype for this example. Whereas the B3 zinc blende structure is maintained for a wide range of pressures and temperatures, we have found [42] that the standard polarity, Bþ0:85P 0:85 under ambient conditions, undergoes a reversal on application of hydrostatic pressure. The inversion occurs through an intermediate situation in which the P valence shell is transferred to a nonnuclear maximum before being caught by the B atom. In this way the low-pressure boron phosphide phase is transformed into high-pressure phosphorus boride, even though both are structurally equivalent.
8.4
Topological Classification of Crystals
The main topological properties of a prototypical set of compounds are listed in Table 8.4. A traditionally debated and often controversial question among solid state scientists is what constitutes the ionicity, covalence, and metallicity of a material. This question can be systematically answered with the aid of the magnitudes provided by the QTAIM analysis [41]. As is apparent from Table 8.4, the electron density at a bond CP, rb, is characteristically large for covalent bonding. It is not easy, however, to compare the bond CP between two di erent pairs of elements in terms of theirs rb values. The Laplacian of the electron density at a point, ‘2rð~rÞ, measures whether the electron density is locally concentrated ð‘2r < 0Þ or depleted ð‘2r > 0Þ there, and provides a detailed map of the basic and acidic regions, respectively, of the crystal [43]. In a typically covalent bond, a region of negative Laplacian would include the bond path together with the two bonded nuclei. Prototypical ionic bonds, in contrast, would have spherical shells of basic character on each nucleus, the bond critical point occurring in a region of acidic character. Between both extreme examples we can find a collection of polar bonding situations with mixed signs. The ratio l1=l3 enables measurement of

2168 Topology and Properties of the Electron Density in Solids
Table 8.4 Topological properties of a representative collection of
crystals, including covalent, ionic, metallic, and molecular compounds. The values rb and ‘2rb are the electron density and the Laplacian, respectively, at a bond CP. l1 al2 al3 are the eigenvalues of the Hessian, and the ratio l1/l3 in the table corresponds to a bond CP. Q(A) is the charge integrated within the basin of A. The last column is the valence density flatness, f (Eq. 3). The results correspond to HF-
LCAO calculations (Section 8.7). All magnitudes are given in atomic units.
Crystal |
AB |
r |
b |
‘2r |
b |
Cl /l |
3 |
A |
Q(A) |
f (%) |
|
|
|
|
1 |
|
|
|
|||
|
|
|
|
|
|
|
|
|
||
Diamond |
CaC |
0.2659 |
0.9044 |
3.75 |
|
C |
0.000 |
4.8 |
||
N2 |
NaN |
0.7380 |
2.9228 |
4.13 |
|
N |
0.000 |
0.0 |
||
|
NaN |
0.0023 |
0.0158 |
0.12 |
|
|
|
|
||
|
NaN |
0.0012 |
0.0099 |
0.10 |
|
|
|
|
||
CaF2 |
CaaF |
0.0297 |
0.1780 |
0.15 |
|
Ca |
1.821 |
1.3 |
||
|
FaF |
0.0127 |
0.0627 |
0.14 |
|
F |
0.911 |
|
||
Li2O |
LiaO |
0.0278 |
0.2055 |
0.15 |
|
Li |
0.897 |
7.3 |
||
|
OaO |
0.0101 |
0.0317 |
0.17 |
|
O |
1.792 |
|
||
Al |
AlaAl |
0.0308 |
0.0084 |
0.47 |
|
Al |
0.000 |
56.8 |
||
Li |
LiaNNM |
0.0071 |
0.0067 |
0.02 |
|
Li |
0.825 |
89.2 |
||
|
NNMaNNM |
0.0078 |
0.0010 |
10.44 |
|
NNM |
0.137 |
|
||
|
|
|
|
|
|
|
|
|
|
|
bond directionality, which is characteristically >1 for covalent bonds and <1 otherwise. Crystal ionicity, on the other hand, can be typically associated with large absolute values of the charge integrated on the basins. Metals are revealed by the flatness of the electron density in the valence region, which can now be measured quantitatively by the ratio of the electron density on the lowest density cage CP to the highest bond CP in the crystal. This flatness index, Eq. (3), can even be particularized to a plane or a line for low-dimensional metallic systems.
Among the crystals in Table 8.4, diamond and N2 clearly have covalent bond CPs. For CaF2 and Li2O there is evidence of large ionicity, and Al and Li are characteristically metallic. For the N2 crystal there is, in addition to the main NaN bond CP, another two types of bond path between neighboring molecules, which are clearly di erentiated by their low density, positive and small Laplacian, and low bond directionality. For the Li crystal, in contrast, nonnuclear maxima of the electron density are observed. The NNM have been shown to occur in many homoatomic molecules and crystals when the appropriate internuclear distance regime is examined [38]. The appropriate distance range is, usually, much smaller than the typical equilibrium distances, and only a few atomic combinations can have NNM under normal thermodynamic conditions. These include metals, for example Li and Na, and also crystals, for example CaC2. The occur-

8.5 Bond Properties – Continuity from the Molecular to the Crystalline Regime 217
rence of NNM is neither characteristic nor exclusive of metals, as was conjectured when this rare topological feature was first discovered.
8.5
Bond Properties – Continuity from the Molecular to the Crystalline Regime
One of the most important features of solid state behavior with regard to the application of QTAIM methods is enlargement of the narrow window of bonding ranges of molecules. In solids, external properties such as pressure and temperature, or the occurrence of polymorphism within a given substance, enable us to observe a given bond over a wide range of di erent distances within the same chemical environment, and even tune these distances at will. Whereas consideration of, for example, the NaN bond in N2 at distances di erent from the equilibrium distance is theoretically easy, this is not achievable in the gas phase, and thus molecular quantum chemists seldom look into the dependence on distance of bond properties. This dependence has, in contrast, been the topic of several solid state studies both of ionic bonds [34, 37, 42, 44] and of intramolecular and intermolecular hydrogen bonds (by Espinosa et al. [45–47] and Ga´lvez et al. [48]). All these studies show that for the same bond (or type of bond) in di erent solidstate conditions there is clear dependence of bond densities ðrbÞ and Laplacians ð‘2rbÞ on interatomic distance, usually linear in a logarithmic plot. Thus, geometric arguments related to ionic radii, for example, should be intimately related to bond properties, and these are, in turn, related to bond energetics [49]. These arguments can, however, be taken a step further; there is, in fact, a continuous bonding regime from the molecular to the crystalline limits [50, 51], with several universal features [52].
To unveil the relationship between the geometrical arguments and the di erent chemical behavior of bonds, it is most useful to seek approximate models that provide simplified, even analytical results. In this respect, the promolecular model (Section 8.7) provides an atomistic image of the chemical bond; using the sum of free atomic densities as an approximation, a good qualitative picture of the bond properties is obtained. This can be seen in Fig. 8.4, in which the main features of a high-level CISD N2 Laplacian are reproduced qualitatively by the promolecular HF Laplacian. This dependence serves also to explain an important universal feature – the bond type, as given by ‘2rb, is also a function of distance [52]. Thus, N2, a covalently bonded molecule at the equilibrium distance (2.1 bohr) and in a certain range around it, has a positive Laplacian for distances larger than 3 bohr, and again for distances smaller than 1 bohr. This is already present in the simplified promolecular model, being related to a balance between the radial curvature and slope of the atomic densities. It is thus an atomic feature: the atomic shell structure is unchanged at large distances, shared at intermediate distances, where the shells interpenetrate substantially, and is completely fused at very short distances, where the K shells of the N atoms start to interact, initially
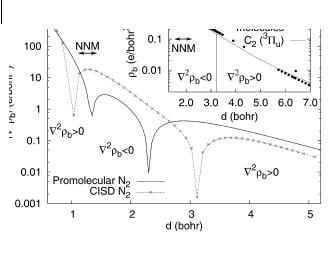
218 8 Topology and Properties of the Electron Density in Solids
Fig. 8.4 Promolecular and CISD//TZVþ(3d,1f ) bond point Laplacians (absolute value, on a logarithmic scale) in the N2 X –1Sþg ground state. The inset shows CaC bond point densities for a set of B3LYP//6- 311G(3df,p) molecules (C2, ethane, ethene, ethine, benzene, anthracene, alene) and PBE-fpLAPW crystals (diamond and graphite at a wide range of distances, plus CaC2), with the bond point density dependence on CaC distance for C2 3Pu.
in a unshared manner. The same kind of behavior can be seen in other homodiatomics, for example Na2 and Ne2, with very di erent kinds of bond at the equilibrium distance, depending mainly on the range at which this distance falls for each of the species. The basic di erence in heterodiatomics with quite di erent electronegativities (NaF and AlN) is that the valence shells do not fuse, but rather are totally or partially transferred from one atom to the other.
Further analysis of the parallel curvature in the bond point enables simple explanation of another important phenomenon – the existence of nonnuclear maxima of the electron density [18, 38]. There is, indeed, a window (marked in the graph) in which, for N2, NNM are observed in the CISD density, and although this is not so for the promolecular density (its curvature falls short of becoming negative by a small amount), it is present for C2 (inset of Fig. 8.4). It can thus be shown to be a feature present in many homonuclear bonds (and also some heteronuclear bonds [42]), and easily related to atomic shell structure.
In addition to this universal behavior of the bond density and Laplacian, with evolution quite similar in di erent compounds, they also have two more universal quantitative features. The first is apparent from analysis of the same type of bond in di erent molecules and crystals – the dependence of the bond properties on distance is the same for a given pair of atoms, continuous even for quite different types of bonding, as explained above. An example is seen in the inset of Fig. 8.4, in which data from single, double, and triple CaC bonds are depicted

8.6 Basin Partition of the Thermodynamic Properties 219
with diamond and graphite crystal bond data for a wide range of distances. There is a general trend (slightly di erent for the long-distance interlayer van der Waals bonds in graphite, poorly described by these calculation) in all these situations which can be almost quantitatively fitted by the dependence of the behavior of the C2 molecule on distance. Obviously, this is an even better model than the promolecule, because CaC bond features are included. Thus, the type of bonding is again a question of distance, which can also be read in the reverse way – the chemistry of the molecule determines the type of bond, and hence a given distance must occur. This interrelationship between geometric features and chemical behavior is behind the success of the geometric arguments previously given.
Finally, there is another kind of universal feature, by which bond properties can be transferred even across di erent bonding pairs with a common atom. To do this, the independent variable cannot be the interatomic distance, because this varies substantially with atomic sizes, but is instead the distance, rA, from the common atom to the bond point. When this is done for a large number of group III nitride clusters, the corresponding B3-phase crystals, nitrogen hydrides, and N2, the plot of bond density against rN plot has a common trend, quite similar to the N2 and promolecular trends [52]. To justify this trend, an exponential tail model of the atomic densities within the promolecular scheme, a further simplification, can be used to prove that, in fact, the homodiatomic and di erent heterodiatomic logarithmic plots di er approximately by a constant. This constant is not large, and depends on the di erent rates of decay of the atomic densities. Thus, the density (and to a lesser extent, its Laplacian) at the bond point is a universal function of the distance to a given atom, except for a small term that depends on the nature of the atom at the other end of the bond.
All of these arguments point in the same direction – the general features of bond properties mainly depend on geometry; because geometry depends, in turn, on bond type and strength, we can conclude that, as empirically proven previously, rb and ‘2rb are good indicators of bond strength and type.
8.6
Basin Partition of the Thermodynamic Properties
In addition to a topological theory of chemical bonding, QTAIM is also a very successful atomic quantum theory – its definition of the atom as a proper open system, fulfilling atomic versions of the key theorems of quantum mechanics, provides a starting point for the following discussion. As is well known [49], QTAIM definition of atomic properties entails construction of appropriate local densities for the property at hand, and integrating these within the atomic basin,
OA ¼ WA |
rO |
~rÞ d~r. This |
is termed an atomic average for the property, although |
the nameÐ |
is |
ðmisleading |
– it is a sum of property O values at di erent points, not |
an |
average of values of the property. Then, for properties conforming with this, |
an |
atomic partition can be obtained, O ¼ PA OA. Properties such as atomic |

|
|
8.6 |
Basin Partition of the Thermodynamic Properties |
221 |
||||
Table 8.5 Local bulk moduli (BW, in GPa) and volume fractions ðwWÞ for |
|
|
|
|
||||
|
|
|
|
|||||
some AB2O4 spinels. Note that each wW includes the multiplicity of W |
|
|
|
|
||||
within the crystal unit cell. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Spinel |
wA |
BA |
wB |
BB |
wO |
BO |
||
|
|
|
|
|
|
|
|
|
MgAl2O4 |
0.089 |
282 |
0.099 |
332 |
0.813 |
202 |
|
|
MgGa2O4 |
0.088 |
261 |
0.164 |
284 |
0.749 |
196 |
|
|
ZnAl2O4 |
0.136 |
246 |
0.095 |
335 |
0.769 |
203 |
|
|
ZnGa2O4 |
0.135 |
241 |
0.158 |
308 |
0.707 |
196 |
|
|
|
|
|
|
|
|
|
|
|
Now, by obtaining VðpÞ from a series of aiPI calculations (Section 8.7), for a range of volumes, followed by fitting of an analytical equation of state, and doing the same for the QTAIM-obtained VA values at each V (thus implicitly obtaining VAðpÞ), we can evaluate local compressibilities, kA, and the weighting factors, wA ¼ VA=V [19]. This leads to the results gathered in Table 8.5.
From these results, the total B values are 216, 211, 216, and 214 GPa, with numerical fitting errors smaller than 1 GPa on each. Now, the reasons behind the BA200 GPa constant value are clear from Eq. (5) – the larger contributions to the compressibility will be those with larger wA and larger 1=BA, both cases pointing to the O atoms. For the volume fraction, not only is the O atom, anion-like, larger than any of the cation-like atoms, but also there are four of them per sevenatom formula unit and hence wO contributes more than 70% in all crystals. For the local compressibility/inverse local bulk modulus, anion-like O atoms are more compressible than the hard, positively charged, A and B cation-like atoms, but the di erence is not as large: BO is approximately 200 GPa, divalent Mg and Zn have BA values of approximately 270 and 245 GPa, and trivalent Al and Ga have BB of approximately 335 and 290 GPa. Thus the average will be clearly dominated by the O atom, which has a remarkably similar compressibility in all four spinels considered, and is expected to behave in the same way in other spinels.
These arguments are general in their spirit but particular in their values. For example, the 20 alkali halides in the B1 phase [19] have quite di erent bulk moduli, ranging from 7.6 GPa in CsI to 80.9 GPa in LiF. The reason for this is mainly the large variation in sizes, with cations taking from less than 8% (LiI) to almost 70% (CsF) of the unit cell volume. There is also a large variability of local BA, both for di erent atoms (36, 35, 20, 18, and 12 GPa for the cation in the ACl series) and for the same atom in di erent compounds (31, 27, 14, 13, and 9 for the Cl atom in the same ACl series). This is because of the much softer regions of the local VAðpÞ equation of state sampled by these compounds, compared with the hard spinels – divalent and trivalent cations are very small and incompressible, and the stronger electrostatic forces also place a large stress over the O atom, whereas the comparatively larger monovalent cations are much more com-