

Matta, Boyd. The quantum theory of atoms in molecules
.pdf232 9 Atoms in Molecules Theory for Exploring the Nature of the Active Sites on Surfaces
ated to ‘rðrÞ ¼ 0. The NR algorithm requires evaluation of the first and second partial derivatives of r, at arbitrary points r.
The points of the necessary gradient paths to determine the bond paths, crystal graphs, and IAS are solutions of the di erential equation [1]:
drðsÞ=ds ¼ ‘rðrðsÞÞ |
ð2Þ |
where the notation rðsÞ implies that a point r on a given path is dependent upon the path parameter s. Equation (2) represents three first-order di erential equations (dxiðsÞ=ds ¼ qr=qxi, xi ¼ x; y; z) and yields unique solutions only when particular values are assigned to three constants of integration. This corresponds to fixing some initial point on a trajectory, at s ¼ s1, for example. A trajectory of the gradient vector field of r(r) is a parametrized integral curve, a solution curve, of the di erential equation for ‘r(r). By fixing a point on a given trajectory all other points which lie on the same path, can be obtained by solving Eq. (2). This is achieved by using a fifth-order Cash–Karp Runge–Kutta (CKRK) method [12]. The general form of the Runge–Kutta formula is:
xnþ1 ¼ xn þ c1k1 þ c2k2 þ c3k3 þ c4k4 þ c5k5 þ c6k6 |
ð3Þ |
where xn ¼ ðxn; yn; znÞ and kj over an interval h are:
k1 ¼ h‘rðrÞjr¼xn
k2 ¼ h‘rðrÞjr¼xn þb21k1
k6 ¼ h‘rðrÞjr¼xn þb61k1 þ þb65k5
The particular values of the various constants ðcj; bijÞ are given in Ref. [12].
It is apparent the key for implementation of the NR and CKRK algorithms is calculation of the required derivatives of r(r), at arbitrary points r. To develop a method enabling us to study complex systems, irrespective of the basis set (analytically or numerically) used, a numerical method on electron densities given on regular, not necessarily homogeneous three-dimensional grids was implemented. The necessary partial derivatives are evaluated using a five-degree Lagrange polynomial interpolation of r(r) and are fed into an automated algorithm for systematic determination of the all CPs. For just one dimension the interpolating polynomial of degree n 1 through n points y1 ¼ f ðx1Þ, y1 ¼ f ðx1Þ; . . . ; yn ¼ f ðxnÞ is given by the Lagrange’s formula:
|
|
|
n |
|
|
n |
|
j |
Y1; j0kðx xjÞ |
|
|
PðxÞ ¼ Xk 1 |
|
|
¼ |
yk |
ð4Þ |
|
|
n |
|||
¼ |
j |
|
Y1; j0kðxk xjÞ |
|
|
|
|
¼ |
|
|
|

9.2 Implementing the Determination of the Topological Properties of rðrÞ |
233 |
|
There are n terms, each a polynomial of degree n 1 and each constructed to be zero at all xj except one, at which it is constructed to be yk. For a homogeneous grid, xj ¼ x1 þ ð j 1Þh, where h is the step size. Defining s ¼ ðx xaÞ=h so that xa and xaþ1 are the central points of the grid, we obtain x ¼ xa þ sh. Substituting this last expression in Eq. (4) we have:
|
|
|
|
n |
|
|
|
|
|
n |
j |
|
Y1; j0kða j þ sÞ |
|
|
|
|||
P ¼ Xk 1 |
|
¼ |
|
|
yk |
ð5Þ |
|||
|
|
|
n |
|
|||||
¼ |
|
|
j |
Y1; j0kðk jÞ |
|
|
|
||
|
|
|
|
¼ |
|
|
|
|
|
and |
|
|
|
|
|
|
|
|
|
n |
|
|
|
|
|
|
|
|
|
P ¼ Xk 1 wk; nðsÞ yk |
|
|
ð6aÞ |
||||||
¼ |
|
|
|
|
|
|
|
|
|
where |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
n |
|
|
|
|
|
|
|
j |
Y1; j0kða j þ sÞ |
|
||||
wk; nðsÞ ¼ |
|
¼ |
|
|
|
ð6bÞ |
|||
|
|
n |
|
|
|||||
|
|
|
|
j |
Y1; j0kðk jÞ |
|
|
|
|
|
|
|
|
|
¼ |
|
|
|
|
wk; nðsÞ are polynomials of degree n 1 in s. The n-degree derivative of these expressions is:
dvP |
|
1 |
|
n |
|
|||
|
|
¼ |
|
Xk 1 wkðv; ÞnðsÞ yk |
|
|||
dx v |
hv |
|
||||||
|
|
|
|
|
¼ |
|
ð7Þ |
|
wðvÞ |
|
|
|
dvwk; nðsÞ |
||||
s |
|
|
|
|||||
k; n |
ð |
Þ ¼ |
|
dsv |
|
|
||
Equations (6) and (7) provide an accurate, rapid, and e cient way of interpolating r(r) and its derivatives at many arbitrary points. The easiest way to determine those derivatives is to develop wk; nðsÞ as an s polynomial. For example, in Table 9.1 the s polynomial expression for wk; nðsÞ and its first and second derivatives for interpolation with n ¼ 4; 6, and 8 points are reported. The explicit expression for n ¼ 4 (Fig. 9.1) is given by:
rðxÞ ¼ ð s3 þ 3s2 2sÞ=6r1 þ ðs3 2s2 s þ 2Þ=2r2 |
|
þ ð s3 þ s2 þ 2sÞ=2r3 þ ðs3 sÞ=6r4 |
ð8Þ |
For a three-dimensional system P is evaluated at points surrounding a box containing the current point and in such a way that any coordinates of the point



236 9 Atoms in Molecules Theory for Exploring the Nature of the Active Sites on Surfaces
values on a plane that coincides with just one of the coordinates of the current point (open circle) is determined. Then, on the plane, a linear mesh (Fig. 9.2c) that coincides with an additional coordinate of the current point is approximated. Finally, the value of P is interpolated on this linear arrangement.
The final equation is:
n1 |
n2 |
n3 |
|
P ¼ kX1 1 kX2 1 kX3 1 wk1; n1 ðs1Þ wk2 ; n2 ðs2Þ wk3; n3 ðs3Þ yk1k2k3 |
ð9Þ |
||
¼ |
¼ |
¼ |
|
Taking advantage of the crystal symmetry and the properties of the gradient paths of r(r) associated with the CPs, we have implemented a very rapid, automated algorithm for systematic determination of the all CPs inside the unit cell of the crystal. First, it determines the bond CP among each pair of atoms (first and second neighbors) using the NR method. Because the gradient paths associated with the negative eigenvalues at the bond CPs originate mainly in the cage CPs, the algorithm then simply searches the origin of some of these paths (for each bond CP) applying the NR method at these points. Finally, searching along the lines connecting nearest neighbor cage CPs, all the ring CPs can be found. Symmetry is used to generate all the CPs having the same type of symmetry (same Wycko letter) and to avoid calculation of a CP that has already been determined.
9.3
An Application to Nanocatalyts – Exploring the Structure of the Hydrodesulfurization MoS2 Catalysts
Transition metal sulfides (TMS) are a very important class of catalysts characterized by stability under harsh conditions in hydrodesulfurization (HDS), hydrodenitrogenation (HDN), and hydrogenation reactions [13–15]. In these processes the surfaces of the sulfides are reduced by sulfur elimination, by use of a large excess of hydrogen at temperatures ranging from 573 to 673 K creating coordinatively unsaturated sites (CUS) or vacancies around the metals. The CUS behave as electron-withdrawing sites whose properties may be regarded as a Lewis acid type center interacting with electron-donating organic substrates [16–18]. It is suggested the nature of these sites is intimately related to the metal–sulfur bond strength [16–19]. Basic studies support the idea that di erences between catalytic activity is related to variations in the concentration of CUS (the Lewis acid sites), which in turn depend on the metal–sulfur bond strength [20–31]. Nickel (and cobalt)-promoted molybdenum sulfide catalysts have for many years been regarded as being among the most important catalysts used in refineries. Studies using X-ray absorption fine structure (EXAFS) have established that the active Mo atom is present as small MoS2-like nanostructures [32, 33]. Adsorption and activity experiments [34, 35] have revealed that the active sites reside at the edges

9.3 An Application to Nanocatalyts 237
of the MoS2 structures, and high-resolution scanning tunneling microscopy (STM) and density-functional theory (DFT) studies [36] have recently shown that the MoS2 nanoclusters adopt a hexagonal shape exposing two di erent types of edge, Mo edges covered with S monomers and fully sulfur-saturated S edges. Incorporation of nickel or cobalt into these edges significantly increases the activity of the catalyst [14, 37–39]. Under typical sulfidation conditions Ni is preferentially incorporated into the metal edge.
9.3.1
Catalyst Models
MoS2 bulk is a layered-type crystal the lattice of which is described by the hexagonal space group P63/mmc with a ¼ b ¼ 3.160 A˚ and c ¼ 12.294 A˚ [40]. Its crystal structure belongs to a family of polytypic structures with close-packed triangular double layers of S with Mo atoms arranged in the trigonal–prismatic holes of the S double layers (Fig. 9.3).
Mo atoms occupy the 2c Wyckof positions with coordinates (1/3, 2/3, 1/4) and the S atoms the 4f position with coordinates (2/3, 1/3, 0.371) [41]. Each Mo atom is bonded to six S atoms in a trigonal–prismatic arrangement. The closest SaS distances are across the double layer and within the close-packed layers; the interlayer SaS distances are much larger and of the van der Waals type. The morphology of the catalysts [16] can be depicted as small MoS2 particles (crystallites) dispersed at the surface of the support (usually SiO2, graphite, etc). These particles have an average size of approximately 600 G200 A˚ 2 and their reactivity depends on preferential exposed faces or planes. HDS catalysis is largely a surface process and, therefore, we must consider surface models. These models are usually obtained by cutting the three-dimensional bulk structure along a particular plane defined by using the appropriate Miller index [29, 39, 42]. For example, the so called basal plane of a MoS2 crystallite [36] is produced by cleaving the crystal along the (001) plane (Figs. 9.3b and 9.3c). This plane is fully covered by sulfur atoms and is inactive for HDS reactions. Cleavage of the bulk structure parallel to (010) plane (Figs. 9.3d and 9.3e) produces the well known edge surface exposing coordinatively unsaturated molybdenum or sulfur atoms. Each of the exposed Mo atoms is coordinated to four sulfur atoms and each terminal sulfur atom is coordinated to two Mo atoms. Several studies have shown that the bare Mo edge terminating in a row of undercoordinated Mo atoms is very unfavorable; such edges will therefore have high a nity for S adsorption. The real morphology of the MoS2 catalyst active sites has been deduced from experimental and theoretical studies. STM enables direct imaging of catalytically relevant surface structure on the atomic scale. By studying a realistic HDS model system consisting of a few-nanometer-wide gold-supported MoS2 particles it has been shown that the morphology of the nanoparticles is sensitive to sulfiding and reaction conditions [36]; this means triangles are formed under heavy sulfiding conditions and truncated hexagons under more sulfo-reductive conditions resembling HDS conditions. These hexagonal clusters expose the basal plane and two di erent

238 9 Atoms in Molecules Theory for Exploring the Nature of the Active Sites on Surfaces


9.3 An Application to Nanocatalyts 241
9.3.2
The Full r(r) Topology of the MoS2 Bulk
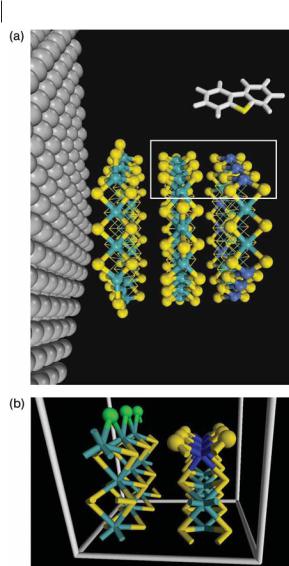
In the MoS2 unit cell, six sulfur atoms locally coordinate with one Mo atom to form a trigonal prismatic structure. Each Mo atom is surrounded by the six nearest sulfur atoms at a distance of 2.404 A˚ forming an MoS2 sheet, and each S atom is surrounded by the three nearest Mo atoms on the sheet and by the three second nearest S atoms located on a neighboring MoS2 sheet. All the CPs of that unit cell have been located and the data that characterize them are given in Table 9.2. In this table the corresponding Wycko letter in the International Tables of Crystallography [57] identifies the critical points within a unit cell. This identification is useful for determining the topology of the electron density of an extended system. Figure 9.5a illustrates the bond and cage CPS determined inside the MoS2 sheets. The bond paths are shown as gray lines connecting the bound atoms. There are six nuclei in the primitive cell, two molybdenum (light blue spheres), located at the position labeled c, and four sulfur (yellow spheres), at position f. There are twelve MoaS bond critical points (gray spheres) at position k, six four-membered ring CPs at f , and one trigonal prism-like cage (red spheres) at b.
Table 9.2 |
Topological properties (au) of r(r) at the critical points for |
|
|
|||
MoS2 bulk space group: P63/mmc (D4 in Schoenflies notation). |
|
|
||||
|
|
6h |
|
|
|
|
|
|
|
|
|
|
|
Wycko |
Site |
Critical point |
l1 |
l2 |
l3 |
rb |
letter |
symmetry |
|
|
|
|
|
|
|
|
|
|
|
|
k 12 |
CS |
MoaS b |
0.1073 |
0.0809 |
0.3212 |
0.0908 |
g 6 |
C2h |
SaS b |
0.0056 |
0.0056 |
0.0388 |
0.0112 |
h 6 |
C2v |
Four-membered |
0.0347 |
0.0058 |
0.0492 |
0.0433 |
|
|
2Moa2S r |
|
|
|
|
b 2 |
D3h |
Three-membered |
0.0149 |
0.0288 |
0.0292 |
0.0159 |
|
|
3 Mo r |
|
|
|
|
k 12 |
CS |
Four-membered |
0.0024 |
0.0062 |
0.0127 |
0.0070 |
|
|
1Moa3S r |
|
|
|
|
d 2 |
D3h |
Red c (Fig. 9.6b) |
0.0253 |
0.0260 |
0.0664 |
0.0311 |
f 4 |
C3v |
Green c (Fig. 9.6c) |
0.0048 |
0.0062 |
0.0062 |
0.0057 |
a 2 |
D3d |
Pink c (Fig. 9.6e) |
0.0017 |
0.0022 |
0.0022 |
0.0045 |
c 2 |
D3h |
Mo n |
|
|
|
|
f 4 |
C3v |
S n |
|
|
|
|